
Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis

 ,
, 
Abstract
:Simple Summary
Abstract
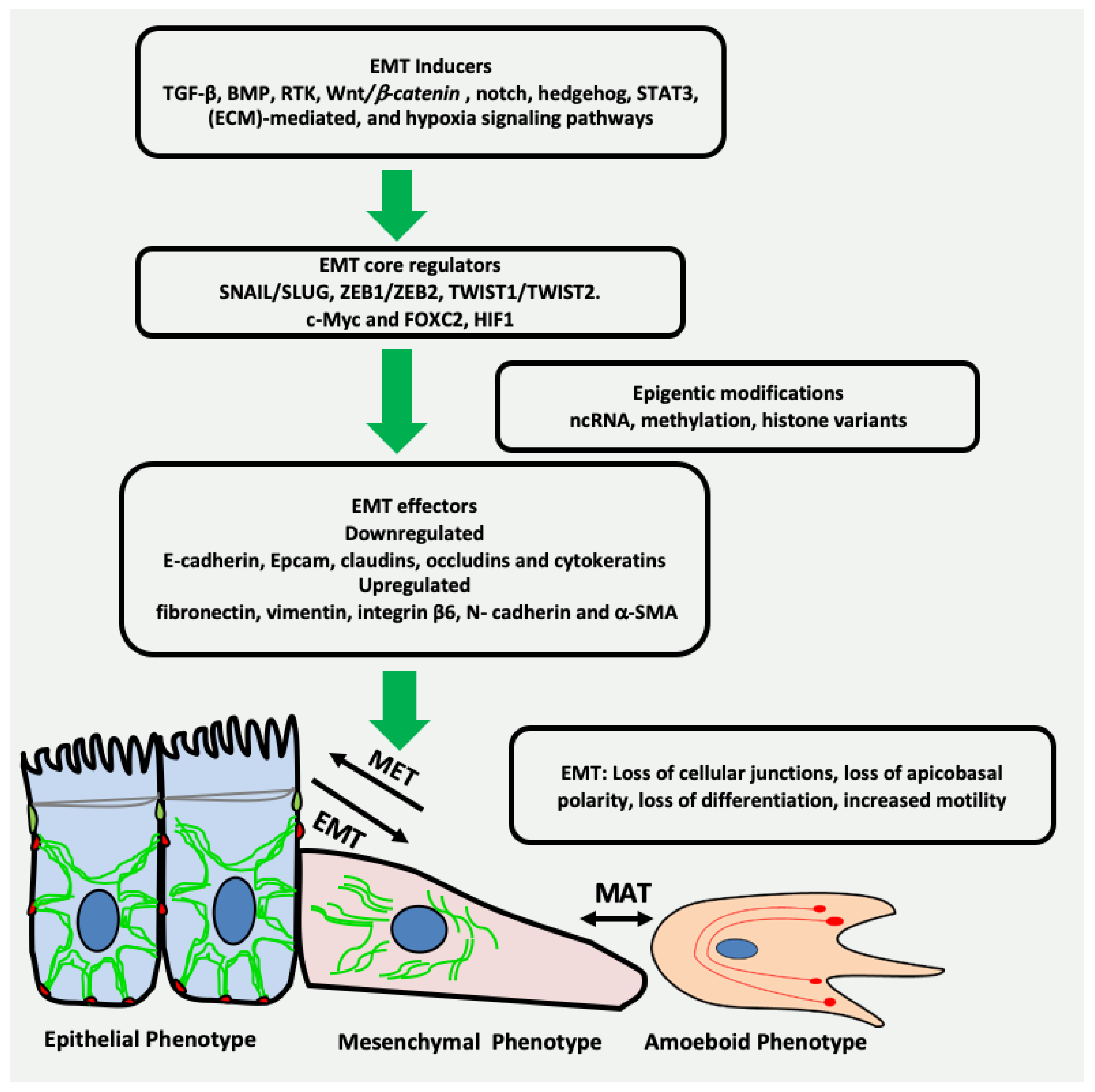
1. Introduction
2. EMT May Produce Cancer Stem Cells (CSCs) Expressing Vimentin
3. Vimentin Expression during Mesenchymal–Amoeboid Transition (MAT) of CSC
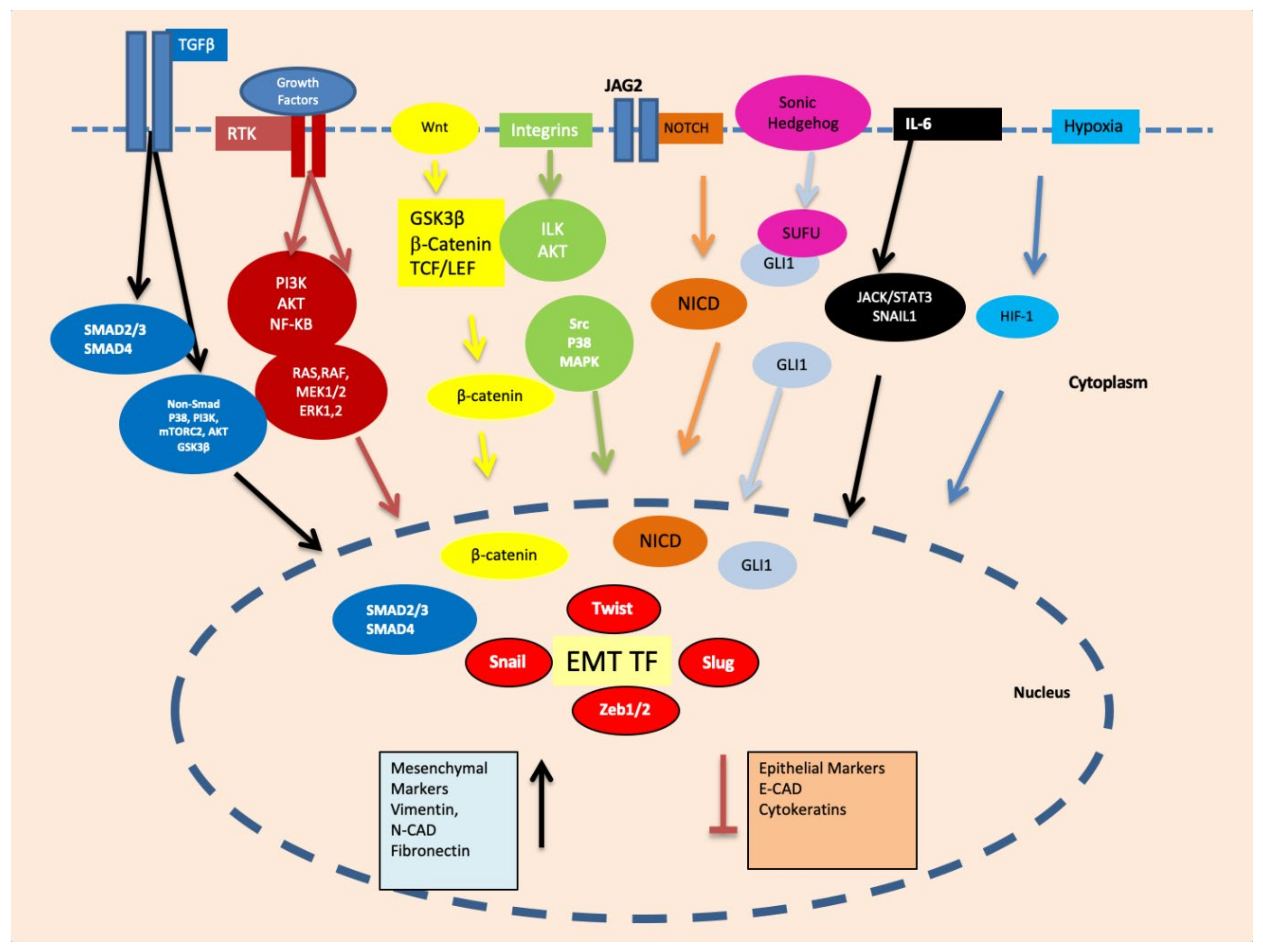
4. Transcription Factors Inducing EMT
4.1. Snail and Slug
4.2. TWIST1
4.3. ZEB1 and ZEB2
4.4. c-MYC
4.5. HIF-1
5. Regulation of Vimentin by Epigenetic Factors
5.1. DNA Methylation
5.2. MicroRNAs and Non-Coding RNAs
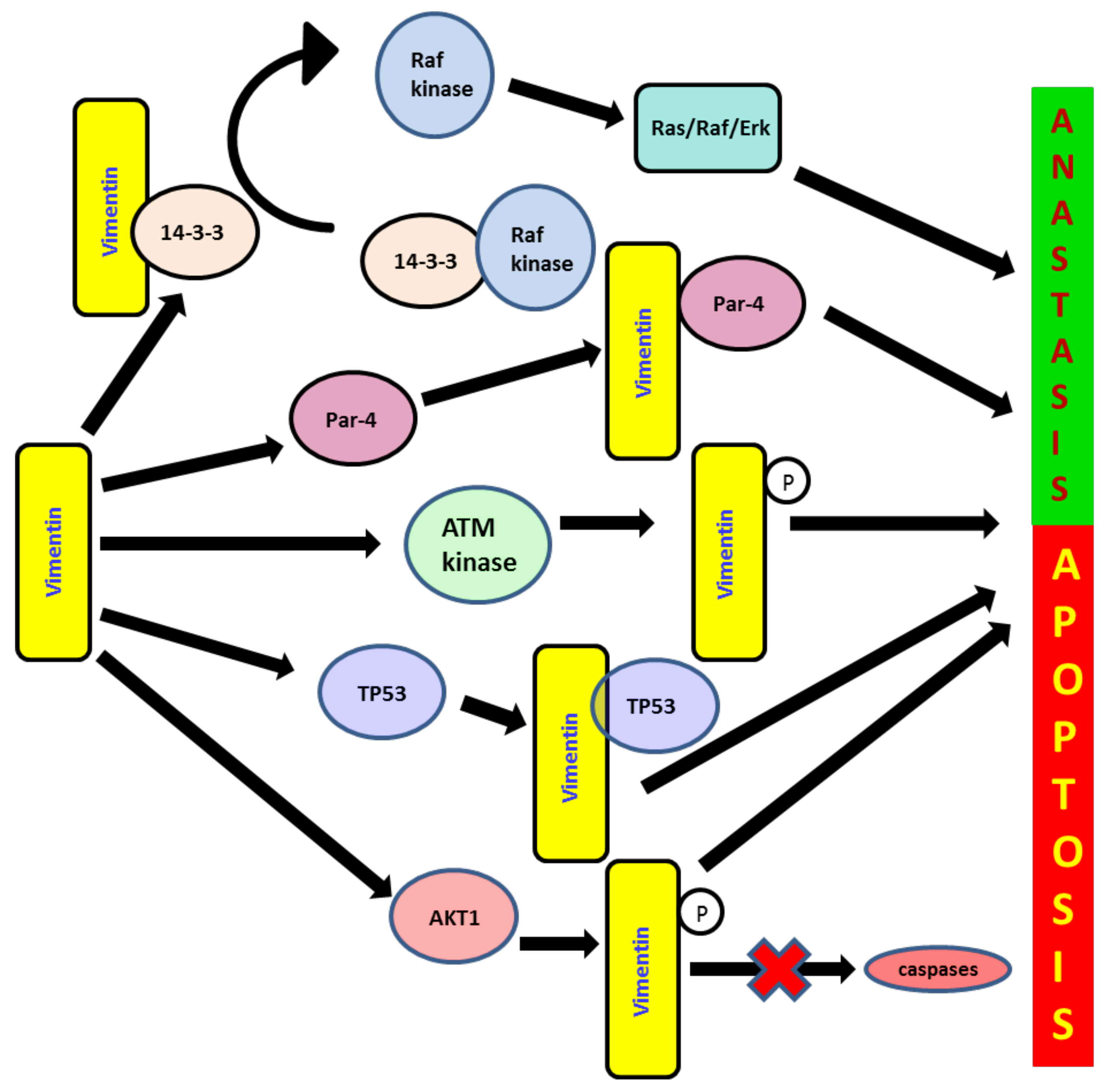
6. Role of Vimentin in Anoikis and Anastasis
7. Role of Vimentin in the DNA Repair System during EMT
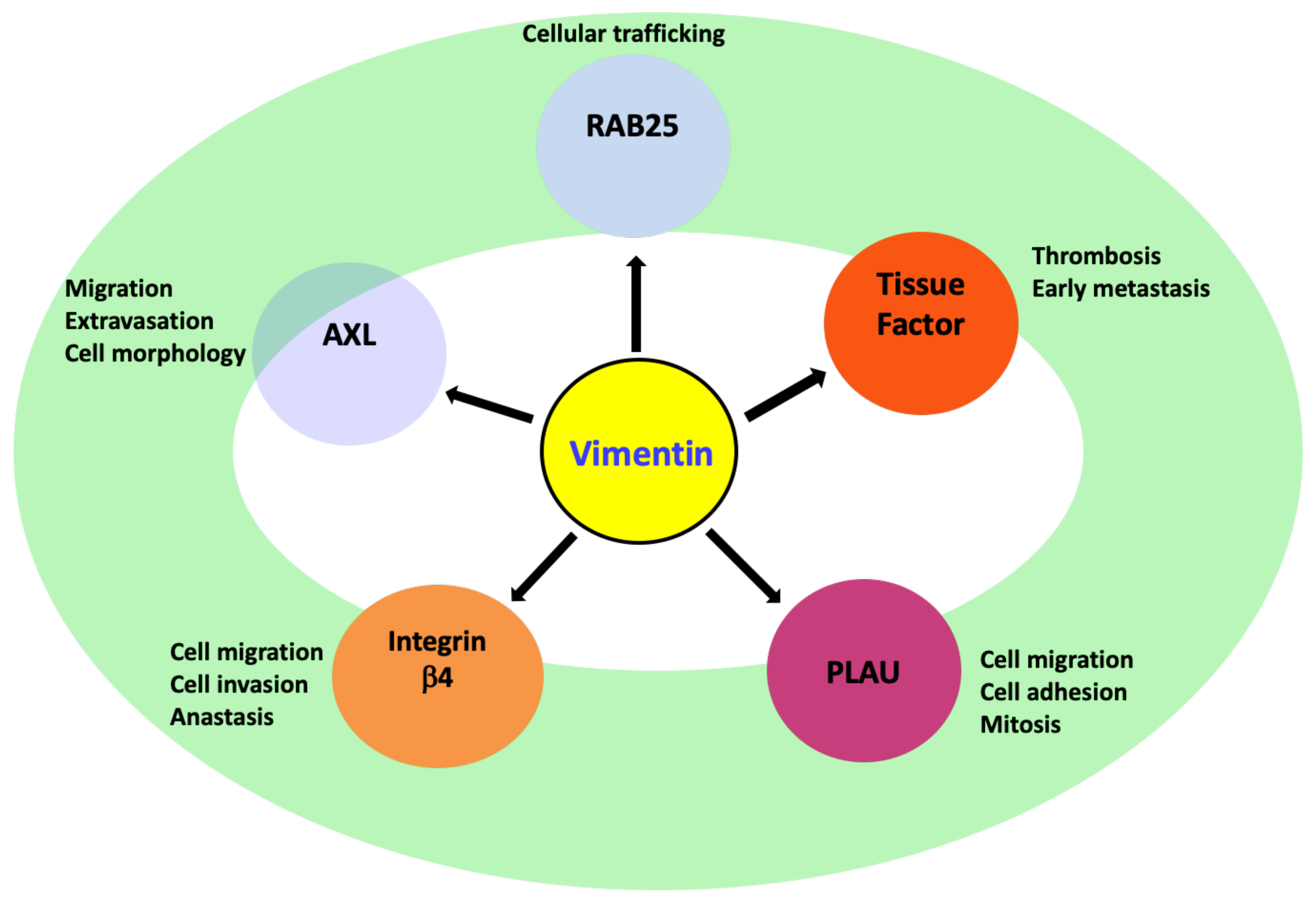
8. Vimentin Regulating Other Genes during EMT
8.1. AXL
8.2. Integrin β4 (ITGβ4)/CD104
8.3. PLAU
8.4. Rab-25
8.5. Tissue Factor (TF)
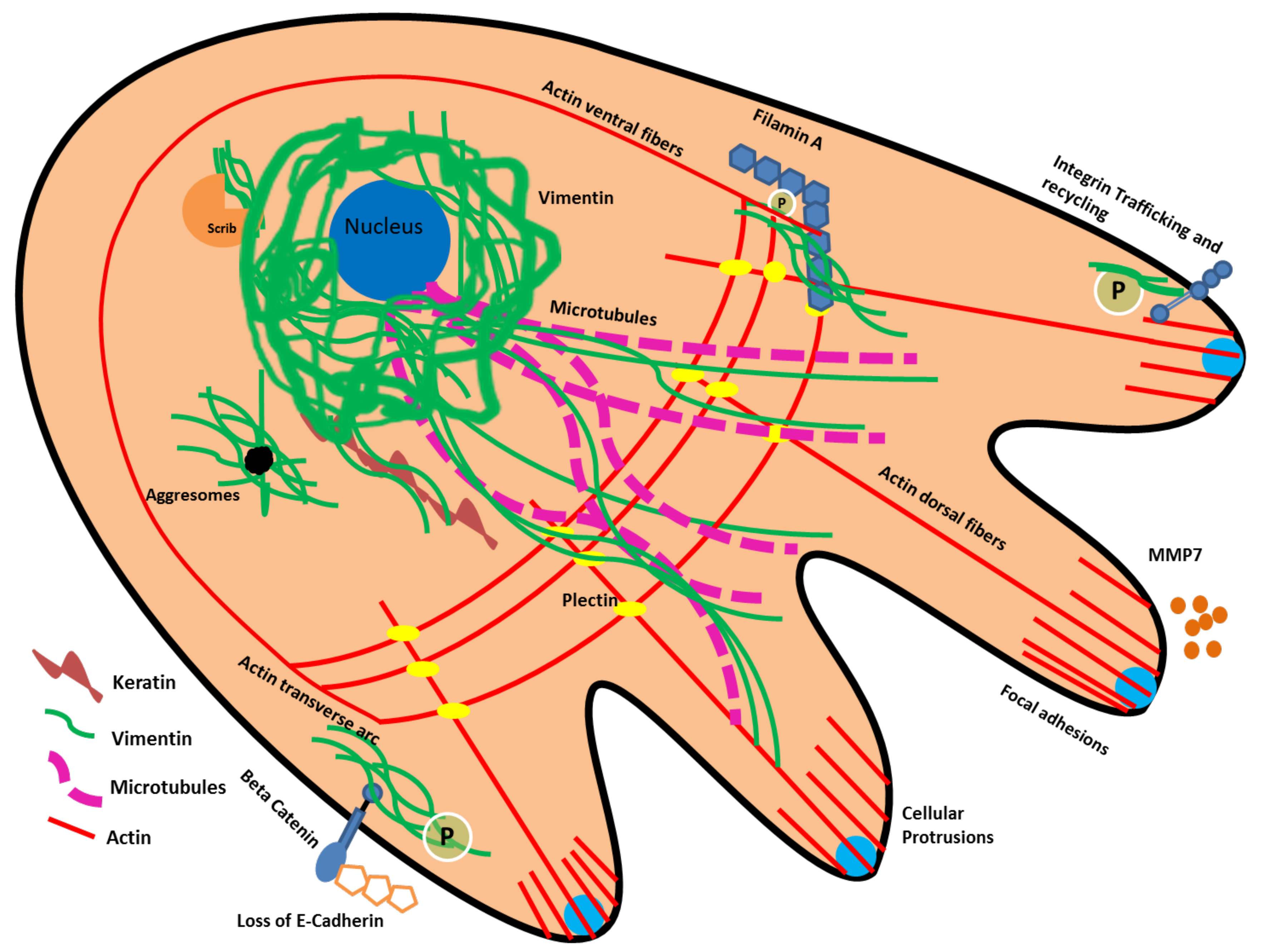
9. Vimentin Protein Interactions and Cytoskeletal Reorganization Related to EMT
10. Effect of Microbial Pathogens and Chronic Inflammation on EMT
11. Conclusions and Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Rout-Pitt, N.; Farrow, N.; Parsons, D.; Donnelley, M. Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir. Res. 2018, 19, 1–10. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, N.V.; Johnson, G.L.; Abell, A.N. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 2011, 10, 2865–2873. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2018, 194, 161–184. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2018, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [Green Version]
- Yeo, C.D.; Kang, N.; Choi, S.Y.; Kim, B.N.; Park, C.K.; Kim, J.W.; Kim, S.J. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: A possible link to epigenetic regulation. Korean J. Intern. Med. 2017, 32, 589–599. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5511947/ (accessed on 2 October 2021). [CrossRef]
- Gonzalez, D.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, S.; Langhans, S.A. Crosstalk of Oncogenic Signaling Pathways during Epithelial–Mesenchymal Transition. Front. Oncol. 2014, 4, 358. [Google Scholar] [CrossRef]
- Georgakopoulos-Soares, I.; Chartoumpekis, D.V.; Kyriazopoulou, V.; Zaravinos, A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Wu, K.-J. Epigenetic regulation of epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β signaling. J. Biomed. Sci. 2020, 27, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Scanlon, C.; Van Tubergen, E.; Inglehart, R.; D’Silva, N. Biomarkers of Epithelial-Mesenchymal Transition in Squamous Cell Carcinoma. J. Dent. Res. 2012, 92, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielsson, F.; Peterson, M.K.; Araújo, H.C.; Lautenschläger, F.; Gad, A.K.B. Vimentin Diversity in Health and Disease. Cells 2018, 7, 147. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [Green Version]
- Ivaska, J. Vimentin. Small GTPases 2011, 2, 51–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.-P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2010, 30, 1436–1448. [Google Scholar] [CrossRef] [Green Version]
- Patteson, A.E.; Vahabikashi, A.; Pogoda, K.; Adam, S.A.; Mandal, K.; Kittisopikul, M.; Sivagurunathan, S.; Goldman, A.; Goldman, R.D.; Janmey, P.A. Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 2019, 218, 4079–4092. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, S.; Azad, G.K.; Amen, T.; Brielle, S.; Park, J.E.; Sze, S.K.; Meshorer, E.; Kaganovich, D. Vimentin protects differentiating stem cells from stress. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Usman, S.; Jamal, A.; Teh, M.-T.; Waseem, A. Major Molecular Signaling Pathways in Oral Cancer Associated With Therapeutic Resistance. Front. Oral Heal. 2021, 1, 15. [Google Scholar] [CrossRef]
- Thankamony, A.P.; Saxena, K.; Murali, R.; Jolly, M.K.; Nair, R. Cancer Stem Cell Plasticity – A Deadly Deal. Front. Mol. Biosci. 2020, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Lleonart, M. The hypoxic microenvironment: A determinant of cancer stem cell evolution. BioEssays 2016, 38, S65–S74. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Ann. Rev. Pathol. Mech. Dis. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martinez-Saez, E.; Prudkin, L.; et al. TGF-β Receptor Inhibitors Target the CD44high/Id1high Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [Green Version]
- Scheel, C.; Eaton, E.N.; Li, S.H.-J.; Chaffer, C.L.; Reinhardt, F.; Kah, K.-J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and Autocrine Signals Induce and Maintain Mesenchymal and Stem Cell States in the Breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, C.; Weinberg, R.A. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int. J. Cancer 2011, 129, 2310–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walcher, L.; Kistenmacher, A.-K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.-R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Zhang, R.; Tu, J.; Liu, S. Novel molecular regulators of breast cancer stem cell plasticity and heterogeneity. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, W.; Li, Y. Stemness-related markers in cancer. Cancer Transl. Med. 2017, 3, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Thapa, R.; Wilson, G. The Importance of CD44 as a Stem Cell Biomarker and Therapeutic Target in Cancer. Stem Cells Int. 2016, 2016, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Patteson, A.E.; Vahabikashi, A.; Goldman, R.D.; Janmey, P.A. Mechanical and Non-Mechanical Functions of Filamentous and Non-Filamentous Vimentin. BioEssays 2020, 42, e2000078. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Zheng, P.; Tang, J.; Liu, Y. CD24: From A to Z. Cell. Mol. Immunol. 2010, 7, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.J.; Fleming, J.M.; Ali, M.A.; Pesesky, M.W.; Ginsburg, E.; Vonderhaar, B.K. Dynamic regulation of CD24 and the invasive, CD44posCD24negphenotype in breast cancer cell lines. Breast Cancer Res. 2009, 11, 14–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell. 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Pearce, D.J.; Taussig, D.; Simpson, C.; Allen, K.; Rohatiner, A.Z.; Lister, T.A.; Bonnet, D. Characterization of Cells with a High Aldehyde Dehydrogenase Activity from Cord Blood and Acute Myeloid Leukemia Samples. Stem Cells 2005, 23, 752–760. [Google Scholar] [CrossRef]
- Wu, A.; Luo, W.; Zhang, Q.; Yang, Z.; Zhang, G.; Li, S.; Yao, K. Aldehyde dehydrogenase 1, a functional marker for identifying cancer stem cells in human nasopharyngeal carcinoma. Cancer Lett. 2013, 330, 181–189. [Google Scholar] [CrossRef]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem Cell Rep. 2013, 2, 78–91. [Google Scholar] [CrossRef]
- Wang, J.; Zeng, H.; Li, H.; Zhang, J.; Wang, S. Roles of sex-determining region Y-box 2 in cell pluripotency and tumor-related signaling pathways. Mol. Clin. Oncol. 2015, 3, 1203–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Chen, M.; Pan, M.-X. Sex determining region Y-box 2 is a prognostic factor for head and neck squamous cell carcinoma: Evidence from 11 published investigations. J. Cancer Res. Ther. 2020, 16, 434. [Google Scholar] [CrossRef]
- Freier, K.; Knoepfle, K.; Flechtenmacher, C.; Pungs, S.; Devens, F.; Toedt, G.; Hofele, C.; Joos, S.; Lichter, P.; Radlwimmer, B. Recurrent copy number gain of transcription factorSOX2and corresponding high protein expression in oral squamous cell carcinoma. Genes Chromosom. Cancer 2010, 49, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Bayo, P.; Jou, A.; Stenzinger, A.; Shao, C.; Gross, M.; Jensen, A.D.; Grabe, N.; Mende, C.H.; Rados, P.V.; Debus, J.; et al. Loss of SOX2 expression induces cell motility via vimentin up-regulation and is an unfavorable risk factor for survival of head and neck squamous cell carcinoma. Mol. Oncol. 2015, 9, 1704–1719. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Du, W.W.; Fang, L.; Shan, S.W.; Qian, J.; Lin, J.; Qian, W.; Ma, J.; Rutnam, Z.J.; Yang, B.B. The Intermediate Filament Vimentin Mediates MicroRNA miR-378 Function in Cellular Self-renewal by Regulating the Expression of the Sox2 Transcription Factor*. J. Biol. Chem. 2013, 288, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Waldeyer, R. Die Entwicklung der Carcinome. Virchows Arch. F. Path. Anat. 1872, 55, 67–159. [Google Scholar] [CrossRef]
- Waldeyer., R. Die Entwicklung der Carcinome. Virchows Arch. F. Path. Anat. 1867, 41, 470–523. [Google Scholar] [CrossRef]
- Enterline, H.T.; Coman, D.R. The ameboid motility of human and animal neoplastic cells. Cancer 1950, 3, 1033–1038. [Google Scholar] [CrossRef]
- Taddei, M.L.; Giannoni, E.; Morandi, A.; Ippolito, L.; Ramazzotti, M.; Callari, M.; Gandellini, P.; Chiarugi, P. Mesenchymal to amoeboid transition is associated with stem-like features of melanoma cells. Cell Commun. Signal. 2014, 12, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emad, A.; Ray, T.; Jensen, T.; Parat, M.; Natrajan, R.; Sinha, S.; Ray, S.P. An epithelial-mesenchymal-amoeboid transition gene signature reveals subtypes of breast cancer progression and metastasis. bioRxiv 2017. Available online: https://www.biorxiv.org/content/10.1101/219410v2.full (accessed on 13 September 2021).
- Wu, J.-S.; Jiang, J.; Chen, B.-J.; Wang, K.; Tang, Y.-L.; Liang, X.-H. Plasticity of cancer cell invasion: Patterns and mechanisms. Transl. Oncol. 2020, 14, 100899. [Google Scholar] [CrossRef]
- Krakhmal, N.V.; Zavyalova, M.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V. Cancer Invasion: Patterns and Mechanisms. Acta Naturae 2015, 7, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talkenberger, K.; Cavalcanti-Adam, E.; Voss-Böhme, A.; Deutsch, A. Amoeboid-mesenchymal migration plasticity promotes invasion only in complex heterogeneous microenvironments. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752. [Google Scholar] [CrossRef]
- Wolf, K.; Mazo, I.; Leung, H.; Engelke, K.; von Andrian, U.H.; Deryugina, E.I.; Strongin, A.Y.; Bröcker, E.-B.; Friedl, P. Compensation mechanism in tumor cell migration: Mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 2003, 160, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Holle, A.; Devi, N.G.K.; Clar, K.; Fan, A.; Saif, M.T.; Kemkemer, R.; Spatz, J.P. Cancer Cells Invade Confined Microchannels via a Self-Directed Mesenchymal-to-Amoeboid Transition. Nano Lett. 2019, 19, 2280–2290. [Google Scholar] [CrossRef]
- Lavenus, S.B.; Tudor, S.M.; Ullo, M.F.; Vosatka, K.W.; Logue, J.S. A flexible network of vimentin intermediate filaments promotes migration of amoeboid cancer cells through confined environments. J. Biol. Chem. 2020, 295, 6700–6709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strouhalova, K.; Přechová, M.; Gandalovičová, A.; Brábek, J.; Gregor, M.; Rosel, D. Vimentin Intermediate Filaments as Potential Target for Cancer Treatment. Cancers 2020, 12, 184. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 1–36. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; Von Der Ohe, J.; Hass, R. Breast Carcinoma: From Initial Tumor Cell Detachment to Settlement at Secondary Sites. BioMed Res. Int. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Stewart, R.L.; O’Connor, K.L. Clinical significance of the integrin α6β4 in human malignancies. Lab. Invest. 2015, 95, 976–986. Available online: https://pubmed.ncbi.nlm.nih.gov/26121317/ (accessed on 13 September 2021). [CrossRef] [PubMed] [Green Version]
- Smith, B.N.; Burton, L.J.; Henderson, V.; Randle, D.D.; Morton, D.J.; Smith, B.A.; Taliaferro-Smith, L.; Nagappan, P.; Yates, C.; Zayzafoon, M.; et al. Snail Promotes Epithelial Mesenchymal Transition in Breast Cancer Cells in Part via Activation of Nuclear ERK2. PLoS ONE 2014, 9, e104987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Olmeda, D.; Moreno-Bueno, G.; Flores, J.M.; Fabra, A.; Portillo, F.; Cano, A. SNAI1 Is Required for Tumor Growth and Lymph Node Metastasis of Human Breast Carcinoma MDA-MB-231 Cells. Cancer Res. 2007, 67, 11721–11731. [Google Scholar] [CrossRef] [Green Version]
- Virtakoivu, R.; Mai, A.; Mattila, E.; De Franceschi, N.; Imanishi, S.; Corthals, G.; Kaukonen, R.; Saari, M.; Cheng, F.; Torvaldson, E.; et al. Vimentin–ERK Signaling Uncouples Slug Gene Regulatory Function. Cancer Res. 2015, 75, 2349–2362. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, J.; Ying, X.; Lin, P.C.; Zhou, B.P. Twist-mediated Epithelial-mesenchymal Transition Promotes Breast Tumor Cell Invasion via Inhibition of Hippo Pathway. Sci. Rep. 2016, 6, 24606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Massagué, J. Epithelial-Mesenchymal Transitions: Twist in Development and Metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Feldker, N.; Ferrazzi, F.; Schuhwerk, H.; Widholz, S.A.; Guenther, K.; Frisch, I.; Jakob, K.; Kleemann, J.; Riegel, D.; Bönisch, U.; et al. Genome-wide cooperation of EMT transcription factor ZEB 1 with YAP and AP -1 in breast cancer. EMBO J. 2020, 39, e103209. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Yu, J.; Zhang, M.; Qin, F.; Lan, X. ZEB1 promotes tumorigenesis and metastasis in hepatocellular carcinoma by regulating the expression of vimentin. Mol. Med. Rep. 2019, 19, 2297–2306. [Google Scholar] [CrossRef] [Green Version]
- Bindels, S.; Mestdagt, M.; Vandewalle, C.; Jacobs, N.; Volders, L.; Noel, A.; Van Roy, F.; Berx, G.; Foidart, J.-M.; Gilles, C. Regulation of vimentin by SIP1 in human epithelial breast tumor cells. Oncogene 2006, 25, 4975–4985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Cheryan, V.T.; Xu, L.; Rishi, A.K.; Reddy, K.B. Myc mediates cancer stem-like cells and EMT changes in triple negative breast cancers cells. PLoS ONE 2017, 12, e0183578. [Google Scholar] [CrossRef] [Green Version]
- Rathje, L.-S.Z.; Nordgren, N.; Pettersson, T.; Ronnlund, D.; Widengren, J.; Aspenstrom, P.; Gad, A.K.B. Oncogenes induce a vimentin filament collapse mediated by HDAC6 that is linked to cell stiffness. Proc. Natl. Acad. Sci. USA 2014, 111, 1515–1520. [Google Scholar] [CrossRef] [Green Version]
- Kidd, M.E.; Shumaker, D.K.; Ridge, K.M. The Role of Vimentin Intermediate Filaments in the Progression of Lung Cancer. Am. J. Respir. Cell Mol. Biol. 2013, 50, 1–6. [Google Scholar] [CrossRef]
- Monteiro-Reis, S.; Lobo, J.; Henrique, R.; Jerónimo, C. Epigenetic Mechanisms Influencing Epithelial to Mesenchymal Transition in Bladder Cancer. Int. J. Mol. Sci. 2019, 20, 297. [Google Scholar] [CrossRef] [Green Version]
- Hass, R.; Von Der Ohe, J.; Ungefroren, H. The Intimate Relationship Among EMT, MET and TME: A T(ransdifferentiation) E(nhancing) M(ix) to Be Exploited for Therapeutic Purposes. Cancers 2020, 12, 3674. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef]
- Saji, S.; Kawakami, M.; Hayashi, S.-I.; Yoshida, N.; Hirose, M.; Horiguchi, S.-I.; Itoh, A.; Funata, N.; Schreiber, S.L.; Yoshida, M.; et al. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene 2005, 24, 4531–4539. [Google Scholar] [CrossRef] [Green Version]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammouz, R.; Kołat, D.; Kałuzińska, Ż.; Płuciennik, E.; Bednarek, A. MicroRNAs: Their Role in Metastasis, Angiogenesis, and the Potential for Biomarker Utility in Bladder Carcinomas. Cancers 2021, 13, 891. [Google Scholar] [CrossRef]
- Bai, J.; Yeh, S.; Qiu, X.; Hu, L.; Zeng, J.; Cai, Y.; Zuo, L.; Li, G.; Yang, G.; Chang, C. TR4 nuclear receptor promotes clear cell renal cell carcinoma (ccRCC) vasculogenic mimicry (VM) formation and metastasis via altering the miR490-3p/vimentin signals. Oncogene 2018, 37, 5901–5912. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; He, C.; Deng, S.; Li, X.; Cui, S.; Zeng, Z.; Liu, M.; Zhao, S.; Chen, J.; Jin, Y.; et al. MiR-548an, Transcriptionally Downregulated by HIF1α/HDAC1, Suppresses Tumorigenesis of Pancreatic Cancer by Targeting Vimentin Expression. Mol. Cancer Ther. 2016, 15, 2209–2219. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-Y.; Lee, C.-H.; Li, Y.-S.; Huang, J.-T.; Lan, S.-H.; Wang, Y.-F.; Lai, W.-W.; Lin, Y.-J.; Liu, H.-S.; Cheng, H.-C. MicroRNA-146a suppresses tumor malignancy via targeting vimentin in esophageal squamous cell carcinoma cells with lower fibronectin membrane assembly. J. Biomed. Sci. 2020, 27, 1–14. [Google Scholar] [CrossRef]
- Hu, H.-F.; Xu, W.W.; Zhang, W.-X.; Yan, X.; Li, Y.-J.; Li, B.; He, Q.-Y. Identification of miR-515-3p and its targets, vimentin and MMP3, as a key regulatory mechanism in esophageal cancer metastasis: Functional and clinical significance. Signal Transduct. Target. Ther. 2020, 5, 1–13. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, Y.S.; Yun, N.H.; Shin, C.H.; Hong, H.K.; Kim, H.H.; Cho, Y.B. MicroRNA-17-5p regulates EMT by targeting vimentin in colorectal cancer. Br. J. Cancer 2020, 123, 1123–1130. [Google Scholar] [CrossRef]
- Lin, S.-L.; Lin, Y.-H.; Chi, H.-C.; Lin, T.-K.; Chen, W.-J.; Yeh, C.-T.; Lin, K.-H. A Novel Long Non-Coding RNA-01488 Suppressed Metastasis and Tumorigenesis by Inducing miRNAs That Reduce Vimentin Expression and Ubiquitination of Cyclin E. Cells 2020, 9, 1504. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.-W.; Yang, Z.-Y.; Xiang, H.-G.; Bao, R.; Ye, Y.-Y.; Ren, T.; Wang, X.-F.; Shu, Y.-J. MicroRNA-1275 inhibits cell migration and invasion in gastric cancer by regulating vimentin and E-cadherin via JAZF1. BMC Cancer 2019, 19, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Tong, S.; Cui, R.; Fan, J.; Liu, C.; Lin, Y.; Tang, J.; Xie, H.; Lin, P.; Zheng, T.; et al. Long Non-Coding MALAT1 Functions as a Competing Endogenous RNA to Regulate Vimentin Expression by Sponging miR-30a-5p in Hepatocellular Carcinoma. Cell. Physiol. Biochem. 2018, 50, 108–120. [Google Scholar] [CrossRef]
- Dong, Y.; Zheng, Y.; Wang, C.; Ding, X.; Du, Y.; Liu, L.; Zhang, W.; Zhong, Y.; Wu, Y.; Song, X. MiR-876-5p modulates head and neck squamous cell carcinoma metastasis and invasion by targeting vimentin. Cancer Cell Int. 2018, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, J.; Chen, X.; Xu, X.; Cao, G.; Li, H.; Wu, T. LncRNA FTX sponges miR-215 and inhibits phosphorylation of vimentin for promoting colorectal cancer progression. Gene Ther. 2018, 25, 321–330. [Google Scholar] [CrossRef]
- Meng, J.; Chen, S.; Han, J.-X.; Qian, B.; Wang, X.-R.; Zhong, W.-L.; Qin, Y.; Zhang, H.; Gao, W.-F.; Lei, Y.-Y.; et al. Twist1 Regulates Vimentin through Cul2 Circular RNA to Promote EMT in Hepatocellular Carcinoma. Cancer Res. 2018, 78, 4150–4162. [Google Scholar] [CrossRef] [Green Version]
- Hiramoto, H.; Muramatsu, T.; Ichikawa, D.; Tanimoto, K.; Yasukawa, S.; Otsuji, E.; Inazawa, J. miR-509-5p and miR-1243 increase the sensitivity to gemcitabine by inhibiting epithelial-mesenchymal transition in pancreatic cancer. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Wang, M.; Chen, M.; Huang, Y.; Jiang, J. Upregulation of microRNA-138-5p inhibits pancreatic cancer cell migration and increases chemotherapy sensitivity. Mol. Med. Rep. 2015, 12, 5135–5140. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Cui, H.; Liang, J.; Su, X. Role of MicroRNA-30c in cancer progression. J. Cancer 2020, 11, 2593–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhang, Y.; Sui, Z.; Zhang, Y.; Liu, M.; Tang, H. USP14 de-ubiquitinates vimentin and miR-320a modulates USP14 and vimentin to contribute to malignancy in gastric cancer cells. Oncotarget 2016, 8, 48725–48736. [Google Scholar] [CrossRef] [Green Version]
- Shan, S.; Fang, L.; Shatseva, T.; Rutnam, Z.J.; Yang, X.; Lu, W.-Y.; Xuan, J.W.; Deng, Z.; Yang, B.B. Mature MiR-17-5p and passenger miR-17-3p induce hepatocellular carcinoma by targeting PTEN, GalNT7, and vimentin in different signal pathways. J. Cell Sci. 2013, 126, 1517–1530. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.-W.; Wang, H.-W.; Chang, C.-W.; Chu, H.-W.; Chen, C.-Y.; Yu, J.-C.; Chao, J.-I.; Liu, H.-F.; Ding, S.-L.; Shen, C.-Y. MicroRNA-30a inhibits cell migration and invasion by downregulating vimentin expression and is a potential prognostic marker in breast cancer. Breast Cancer Res. Treat. 2012, 134, 1081–1093. [Google Scholar] [CrossRef]
- Shi, Y.; Shi, H.; Zhang, B.; Yan, Y.; Han, X.; Jiang, W.; Qian, H.; Xu, W. miR-373 suppresses gastric cancer metastasis by downregulating vimentin. Mol. Med. Rep. 2017, 17, 4027–4034. [Google Scholar] [CrossRef]
- Wang, T.-H.; Lin, Y.-S.; Chen, Y.; Yeh, C.-T.; Huang, Y.-L.; Hsieh, T.-H.; Shieh, T.-M.; Hsueh, C.; Chen, T.-C. Long non-coding RNA AOC4P suppresses hepatocellular carcinoma metastasis by enhancing vimentin degradation and inhibiting epithelial-mesenchymal transition. Oncotarget 2015, 6, 23342–23357. [Google Scholar] [CrossRef]
- Huang, J.-F.; Guo, Y.-J.; Zhao, C.-X.; Yuan, S.-X.; Wang, Y.; Tang, G.-N.; Zhou, W.-P.; Sun, S.-H. Hepatitis B virus X protein (HBx)-related long noncoding RNA (lncRNA) down-regulated expression by HBx (Dreh) inhibits hepatocellular carcinoma metastasis by targeting the intermediate filament protein vimentin. Hepatology 2012, 57, 1882–1892. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta (BBA) Bioenerg. 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, Y.; Chen, F.; Chang, R.; Trivedi, M.; Green, K.J.; Cryns, V.L. Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death Differ. 2001, 8, 443–450. [Google Scholar] [CrossRef]
- Sun, G.; Montell, D.J. Q&A: Cellular near death experiences—what is anastasis? BMC Biol. 2017, 15, 92. [Google Scholar]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.-S.; Rosenblatt, K.; Huang, K.-L.; Lahat, G.; Brobey, R.; Bolshakov, S.; Nguyen, T.; Ding, Z.; Belousov, R.; Bill, K.; et al. Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 2010, 30, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzivion, G.; Luo, Z.; Avruch, J. Calyculin A-induced Vimentin Phosphorylation Sequesters 14-3-3 and Displaces Other 14-3-3 Partners in Vivo. J. Biol. Chem. 2000, 275, 29772–29778. [Google Scholar] [CrossRef] [Green Version]
- Tzivion, G.; Shen, Y.H.; Zhu, J. 14-3-3 proteins; bringing new definitions to scaffolding. Oncogene 2001, 20, 6331–6338. [Google Scholar] [CrossRef] [Green Version]
- Burikhanov, R.; Sviripa, V.M.; Hebbar, N.; Zhang, W.; Layton, W.J.; Hamza, A.; Zhan, C.-G.; Watt, D.S.; Liu, C.; Rangnekar, V.M. Arylquins target vimentin to trigger Par-4 secretion for tumor cell apoptosis. Nat. Chem. Biol. 2014, 10, 924–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Lu, J.; Wang, C.; Xue, X. The prognostic values of the expression of Vimentin, TP53, and Podoplanin in patients with cervical cancer. Cancer Cell Int. 2017, 17, 80. [Google Scholar] [CrossRef]
- Weyemi, U.; Redon, C.E.; Bonner, W.M. H2AX and EMT: Deciphering beyond DNA repair. Cell Cycle 2016, 15, 1305–1306. [Google Scholar] [CrossRef] [Green Version]
- Drápela, S.; Bouchal, J.; Jolly, M.K.; Culig, Z.; Souček, K. ZEB1: A Critical Regulator of Cell Plasticity, DNA Damage Response, and Therapy Resistance. Front. Mol. Biosci. 2020, 7, 36. [Google Scholar] [CrossRef]
- Tolstonog, G.V.; Mothes, E.; Shoeman, R.L.; Traub, P. Isolation of SDS-Stable Complexes of the Intermediate Filament Protein Vimentin with Repetitive, Mobile, Nuclear Matrix Attachment Region, and Mitochondrial DNA Sequence Elements from Cultured Mouse and Human Fibroblasts. DNA Cell Biol. 2001, 20, 531–554. [Google Scholar] [CrossRef]
- Chakraborty, S.; Kumar, A.; Faheem, M.M.; Katoch, A.; Kumar, A.; Jamwal, V.L.; Nayak, D.; Golani, A.; Rasool, R.U.; Ahmad, S.M.; et al. Vimentin activation in early apoptotic cancer cells errands survival pathways during DNA damage inducer CPT treatment in colon carcinoma model. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, S.; Xu, H.; Ferro, T.J.; Rivera, P.X. Poly(ADP-ribose) polymerase-1 regulates vimentin expression in lung cancer cells. Am. J. Physiol. Cell. Mol. Physiol. 2007, 293, L1127–L1134. [Google Scholar] [CrossRef] [Green Version]
- Kotula, E.; Faigle, W.; Berthault, N.; Dingli, F.; Loew, D.; Sun, J.-S.; Dutreix, M.; Quanz, M. DNA-PK Target Identification Reveals Novel Links between DNA Repair Signaling and Cytoskeletal Regulation. PLoS ONE 2013, 8, e80313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Y.; Zheng, Z.; Chen, Y.; Wang, Q.; Zhang, Z.; Deng, H. Downregulation of vimentin expression increased drug resistance in ovarian cancer cells. Oncotarget 2016, 7, 45876–45888. [Google Scholar] [CrossRef] [Green Version]
- Lilienbaum, A.; Paulin, D. Activation of the human vimentin gene by the Tax human T-cell leukemia virus. I. Mechanisms of regulation by the NF-kappa B transcription factor. J. Biol. Chem. 1993, 268, 2180–2188. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, X.; Salmon, M.; Lin, X.; Zehner, Z.E. TGFβ1 regulation of vimentin gene expression during differentiation of the C2C12 skeletal myogenic cell line requires Smads, AP-1 and Sp1 family members. Biochim. Biophys. Acta (BBA) Bioenerg. 2007, 1773, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogel, M.R.; Soni, P.N.; Troken, J.R.; Sitikov, A.; Trejo, H.E.; Ridge, K.M. Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 2011, 25, 3873–3883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rittling, S.R.; Coutinho, L.; Amram, T.; Kolbe, M. AP-1/jun binding sites mediate serum inducibility of the human vimentin promoter. Nucleic Acids Res. 1989, 17, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Gilles, C.; Polette, M.; Mestdagt, M.; Nawrocki-Raby, B.; Ruggeri, P.; Birembaut, P.; Foidart, J.-M. Transactivation of vimentin by beta-catenin in human breast cancer cells. Cancer Res. 2003, 63, 2658–2664. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Tokunaga, E.; Inoue, Y.; Yamashita, N.; Saeki, H.; Okano, S.; Kitao, H.; Oki, E.; Oda, Y.; Maehara, Y. Impact of Expression of Vimentin and Axl in Breast Cancer. Clin. Breast Cancer 2016, 16, 520–526.e2. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Liu, L.-N.; Li, D.-D.; He, Y.-P.; Guo, L.-H.; Sun, L.-P.; Xu, H.-X.; Zhang, X.-P. Integrin β4 promotes cell invasion and epithelial-mesenchymal transition through the modulation of Slug expression in hepatocellular carcinoma. Sci. Rep. 2017, 7, 40464. [Google Scholar] [CrossRef]
- Masugi, Y.; Yamazaki, K.; Emoto, K.; Effendi, K.; Tsujikawa, H.; Kitago, M.; Itano, O.; Kitagawa, Y.; Sakamoto, M. Upregulation of integrin β4 promotes epithelial–mesenchymal transition and is a novel prognostic marker in pancreatic ductal adenocarcinoma. Lab. Investig. 2015, 95, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, R.K.; Varshney, A.; Yadava, P.K. Diversity and functional evolution of the plasminogen activator system. Biomed. Pharmacother. 2018, 98, 886–898. [Google Scholar] [CrossRef]
- Di Mauro, C.; Pesapane, A.; Formisano, L.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Monteleone, F.; et al. Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Zhang, Y.; Zhang, Y.; Xiao, W. Involvement of urokinase in cigarette smoke extract-induced epithelial–mesenchymal transition in human small airway epithelial cells. Lab. Investig. 2015, 95, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Skrypek, N.; Bruneel, K.; Vandewalle, C.; De Smedt, E.; Soen, B.; Loret, N.; Taminau, J.; Goossens, S.; Vandamme, N.; Berx, G. ZEB2 stably represses RAB25 expression through epigenetic regulation by SIRT1 and DNMTs during epithelial-to-mesenchymal transition. Epigenet. Chromatin 2018, 11, 70. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, L.; Li, A.; Han, X. The roles of ZEB1 in tumorigenic progression and epigenetic modifications. Biomed. Pharmacother. 2018, 110, 400–408. [Google Scholar] [CrossRef]
- Bystricky, B.; Reuben, J.M.; Mego, M. Circulating tumor cells and coagulation—Minireview. Crit. Rev. Oncol. 2017, 114, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Ünlü, B.; Versteeg, H.H. Cancer-associated thrombosis: The search for the holy grail continues. Res. Pract. Thromb. Haemost. 2018, 2, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Francart, M.E.; Vanwynsberghe, A.M.; Lambert, J.; Bourcy, M.; Genna, A.; Ancel, J.; Perez-Boza, J.; Noël, A.; Birembaut, P.; Struman, I.; et al. Vimentin prevents a miR-dependent negative regulation of tissue factor mRNA during epithelial-mesenchymal transitions and facilitates early metastasis. Oncogene 2020, 39, 3680–3692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, S.; Viedma-Poyatos, A.; Navarro-Carrasco, E.; Martínez, A.E.; Pajares, M.A.; Pérez-Sala, D. Vimentin filaments interact with the actin cortex in mitosis allowing normal cell division. Nat. Commun. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Cogli, L.; Progida, C.; Bramato, R.; Bucci, C. Vimentin phosphorylation and assembly are regulated by the small GTPase Rab7a. Biochim. Biophys. Acta (BBA) Bioenerg. 2013, 1833, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Nakamura, F.; Lee, W.; Shifrin, Y.; Arora, P.; McCulloch, C.A. Filamin A is required for vimentin-mediated cell adhesion and spreading. Am. J. Physiol. Physiol. 2010, 298, C221–C236. [Google Scholar] [CrossRef] [Green Version]
- Esue, O.; Carson, A.A.; Tseng, Y.; Wirtz, D. A Direct Interaction between Actin and Vimentin Filaments Mediated by the Tail Domain of Vimentin. J. Biol. Chem. 2006, 281, 30393–30399. [Google Scholar] [CrossRef] [Green Version]
- Ramos, I.; Stamatakis, K.; Oeste, C.L.; Pérez-Sala, D. Vimentin as a Multifaceted Player and Potential Therapeutic Target in Viral Infections. Int. J. Mol. Sci. 2020, 21, 4675. [Google Scholar] [CrossRef]
- Velez-Delvalle, C.; Marsch-Moreno, M.; Castro-Muñozledo, F.; Galván-Mendoza, I.J.; Kuri-Harcuch, W. Epithelial cell migration requires the interaction between the vimentin and keratin intermediate filaments. Sci. Rep. 2016, 6, 24389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.; Leube, R.E. Keratin intermediate filaments: Intermediaries of epithelial cell migration. Essays Biochem. 2019, 63, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Dmello, C.; Sawant, S.; Alam, H.; Gangadaran, P.; Mogre, S.; Tiwari, R.; D’Souza, Z.; Narkar, M.; Thorat, R.; Patil, K.; et al. Vimentin regulates differentiation switch via modulation of keratin 14 levels and their expression together correlates with poor prognosis in oral cancer patients. PLoS ONE 2017, 12, e0172559. [Google Scholar] [CrossRef]
- Battaglia, R.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7, 1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Şahin, H.; Güngören, A.; Sezgin, B.; Ün, B.; Şahin, E.A.; Dolapçioğlu, K.; Bayik, R.N. Vascular effect of levonorgestrel intrauterine system on heavy menstrual bleeding: Is it associated with hemodynamic changes in uterine, radial, and spiral arteries? J. Obstet. Gynaecol. 2020, 41, 89–93. [Google Scholar] [CrossRef]
- Gan, Z.; Ding, L.; Burckhardt, C.J.; Lowery, J.; Zaritsky, A.; Sitterley, K.; Mota, A.; Costigliola, N.; Starker, C.G.; Voytas, D.F.; et al. Vimentin Intermediate Filaments Template Microtubule Networks to Enhance Persistence in Cell Polarity and Directed Migration. Cell Syst. 2016, 3, 252–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, C.-Y.; Chai, J.; Tang, T.; Wong, W.; Sethi, G.; Shanmugam, M.; Chong, P.; Looi, C. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.-K.; Jiang, X.-Y.; Zhou, X.-X.; Wang, D.-M.; Song, X.-L.; Jiang, H.-B. Upregulation of vimentin and aberrant expression of E-cadherin/β-catenin complex in oral squamous cell carcinomas: Correlation with the clinicopathological features and patient outcome. Mod. Pathol. 2009, 23, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Ivaska, J.; Vuoriluoto, K.; Huovinen, T.; Izawa, I.; Inagaki, M.; Parker, P.J. PKCepsilon-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 2005, 24, 3834–3845. [Google Scholar] [CrossRef]
- Kim, J.; Jang, J.; Yang, C.; Kim, E.J.; Jung, H.; Kim, C. Vimentin filament controls integrin α5β1-mediated cell adhesion by binding to integrin through its Ser38 residue. FEBS Lett. 2016, 590, 3517–3525. [Google Scholar] [CrossRef]
- Phua, D.C.; Humbert, P.O.; Hunziker, W. Vimentin Regulates Scribble Activity by Protecting It from Proteasomal Degradation. Mol. Biol. Cell 2009, 20, 2841–2855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, H.-I.; Kang, H.; Dave, J.M.; Mendoza, E.A.; Su, S.-C.; Maxwell, S.A.; Bayless, K.J. Calpain-mediated vimentin cleavage occurs upstream of MT1-MMP membrane translocation to facilitate endothelial sprout initiation. Angiogenesis 2012, 15, 287–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, V.A.; Samuel, S.K.; Davie, J. Nuclear matrix proteins associated with DNA in situ in hormone-dependent and hormone-independent human breast cancer cell lines. Cancer Res. 2000, 60, 288–292. [Google Scholar]
- Shoeman, R.L.; Traub, P. The in vitro DNA-binding properties of purified nuclear lamin proteins and vimentin. J. Biol. Chem. 1990, 265, 9055–9061. [Google Scholar] [CrossRef]
- Ceschi, S.; Berselli, M.; Giantin, M.; Toppo, S.; Spolaore, B.; Sissi, C. Vimentin binds to G-quadruplex repeats found at telomeres and gene promoters. bioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.05.25.444966v1 (accessed on 13 September 2021).
- Zhao, C.H.; Li, Q.F. Altered profiles of nuclear matrix proteins during the differentiation of human gastric mucous adenocarcinoma MGc80-3 cells. World J. Gastroenterol. 2005, 11, 4628–4633. [Google Scholar] [CrossRef]
- cBioPortal. Skin Cutaneous Melanoma (TCGA, PanCancer Atlas). 2018. Available online: https://www.cbioportal.org/results?plots_horz_selection=%7B%7D&plots_vert_selection=%7B%7D&plots_coloring_selection=%7B%7D&gene_list=VIM&cancer_study_list=skcm_tcga_pan_can_atlas_2018&case_set_id=all (accessed on 13 September 2021).
- Hofman, P.; Vouret-Craviari, V. Microbes-induced EMT at the crossroad of inflammation and cancer. Gut Microbes 2012, 3, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.K.; Meyer, K.; Di Bisceglie, A.M.; Ray, R.B. Hepatitis C Virus Induces Epithelial-Mesenchymal Transition in Primary Human Hepatocytes. J. Virol. 2012, 86, 13621–13628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.; Gay, C.; Ramkumar, K.; Cargill, K.; Cardnell, R.; Nilsson, M.; Heeke, S.; Park, E.; Kundu, S.; Diao, L.; et al. 1735P SARS-CoV-2 infection induces EMT-like molecular changes, including ZEB1-mediated repression of the viral receptor ACE2, in lung cancer models. Ann. Oncol. 2020, 31, S1015. [Google Scholar] [CrossRef]
- Li, Z.; Paulin, D.; Lacolley, P.; Coletti, D.; Agbulut, O. Vimentin as a target for the treatment of COVID-19. BMJ Open Respir. Res. 2020, 7, e000623. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Pan, P.; Yan, P.; Long, Y.; Zhou, X.; Wang, X.; Zhou, R.; Wen, B.; Xie, L.; Liu, D. Role of vimentin in modulating immune cell apoptosis and inflammatory responses in sepsis. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MicroRNA | Cell Lines | Cancer Type | References |
|---|---|---|---|
| miR-146a | ESCC CE81T cells | Esophageal squamous cell carcinoma | [86] |
| miR-515-3p | KYSE150-Luc-LM3, KYSE410-Luc-I6, KYSE150-Luc,43 and EC109 and NE1-E6E7 | Esophageal squamous cell carcinoma | [87] |
| miR-17-5p | No Access/withdrawn | Colorectal cancer | [88] |
| miR-124-3p/ miR-138-5p | SK-Hep1 and Hep3B | Hepatocellular carcinoma | [89] |
| miR-1275 | MGC803 (CRL-1739) and SGC-7901 (CRL-5822) | Gastric cancer | [90] |
| miR-30a-5p | HepG2, Huh7 and HEK-293T, HCT116 | Hepatocellular carcinoma | [91] |
| miR-876-5p | CAL27 and HEK293T (293T), HNSCC WSU-HN4 and WSU-HN6 | Head and Neck squamous cell carcinoma | [92] |
| miR-490-3p | Caki-1, Sw839, 786-0, A498, OSRC-2, ACHN, 769-P | Clear cell renal cell carcinoma | [84] |
| miR-215 | SW480, HCT116, | Colorectal cancer | [93] |
| miR-1246, | PLC-PRF-5 and SMMC-7721, HEK-293T | Hepatocellular carcinoma | [94] |
| miR-578, miR490-5p | PLC-PRF-5 and SMMC-7721, HEK-293T | Hepatocellular carcinoma | [94] |
| miR-509-5p | Panc1 and KMP3, KP4-4, BxPC3, CFPAC1 and SU.86.86 | Pancreatic cancer | [95] |
| miR-138-5p | AsPC-1, BxPc-3, Capan-1, Capan-2, CFPAC-1, PANC-1, MIA PaCa-2 and SW1990 | Pancreatic cancer | [96] |
| miR-30c | Review article | Global | [97] |
| miR-320a | BGC-823 | Gastric cancer | [98] |
| miR-17-3p | HepG2 | Hepatocellular carcinoma | [99] |
| miR-17-5p | SW480, HT29, LoVo | Colorectal carcinoma | [88] |
| miR-30a | Hs578T and MDA-MB-231 | Breast cancer | [100] |
| miR-378 | U87 | Global | [46] |
| miR-294 | SGC-7901 and HGC-27,MFC | Gastric cancer | [101] |
| AOC4P | J7 and SK-Hep1 | Hepatocellular carcinoma | [102] |
| lncRNA-Dreh | Hepa1-6 | Hepatocellular carcinoma | [103] |
| Cancer Type/Study of Origin | Mutation | Mutation Type | Effect of Mutation on Copy Number | TCGA Sample ID |
|---|---|---|---|---|
| Oligodendroglioma | NPM1-VIM | GENE FUSION | Shallow Deletion | TCGA-FG-6692-01 |
| Dedifferentiated Sarcoma | COL3A1-VIM | GENE FUSION | Diploid | TCGA-DX-A48N-01 |
| Dedifferentiated Sarcoma | COL1A1-VIM | GENE FUSION | Diploid | TCGA-DX-A7EI-01 |
| Myxofibrosarcoma | TNXB-VIM | GENE FUSION | Gain | TCGA-DX-AB2T-01 |
| Cutaneous Melanoma | S100A6-VIM | GENE FUSION | Not known | TCGA-GN-A26C-01 |
| Cutaneous Melanoma | HSPG2-VIM | GENE FUSION | Not known | TCGA-GN-A26C-01 |
| Cutaneous Melanoma | E156K | Missense_Mutation | Not known | TCGA-BF-A5EQ-01 |
| Cutaneous Melanoma | E156K | Missense_Mutation | Gain | TCGA-D3-A3MR-06 |
| Cervical Squamous Cell Carcinoma | E153D | Missense_Mutation | Diploid | TCGA-IR-A3LK-01 |
| Adrenocortical Carcinoma | A247P | Missense_Mutation | Gain | TCGA-OR-A5KB-01 |
| Astrocytoma | M372V | Missense_Mutation | Diploid | TCGA-DU-6392-01 |
| Astrocytoma | E407V | Missense_Mutation | Diploid | TCGA-DU-6392-01 |
| Serous Ovarian Cancer | E407K | Missense_Mutation | Gain | TCGA-59-2348-01 |
| Serous Ovarian Cancer | R320Q | Missense_Mutation | Not known | TCGA-23-2649-01 |
| Serous Ovarian Cancer | R364L | Missense_Mutation | ShallowDel | TCGA-24-1849-01 |
| Serous Ovarian Cancer | N283D | Missense_Mutation | Diploid | TCGA-25-2397-01 |
| Glioblastoma Multiforme | A301T | Missense_Mutation | Diploid | TCGA-06-5416-01 |
| Glioblastoma Multiforme | E95Q | Missense_Mutation | Shallow Deletion | TCGA-14-0866-01 |
| Sarcoma ** | V171F | Missense_Mutation | Diploid | TCGA-X9-A973-01 |
| Lung Squamous Cell Carcinoma | F15L | Missense_Mutation | Diploid | TCGA-21-5782-01 |
| Lung Squamous Cell Carcinoma | R222L | Missense_Mutation | Diploid | TCGA-33-4587-01 |
| Lung Squamous Cell Carcinoma | E244K | Missense_Mutation | Diploid | TCGA-37-3783-01 |
| Lung Squamous Cell Carcinoma | A332S | Missense_Mutation | Shallow Deletion | TCGA-39-5021-01 |
| Lung Squamous Cell Carcinoma | E187G | Missense_Mutation | Shallow Deletion | TCGA-39-5036-01 |
| Lung Squamous Cell Carcinoma | L340M | Missense_Mutation | Shallow Deletion | TCGA-22-4613-01 |
| Lung Squamous Cell Carcinoma | D429Y | Missense_Mutation | Diploid | TCGA-39-5035-01 |
| Bladder Urothelial Carcinoma | E407K | Missense_Mutation | Gain | TCGA-KQ-A41P-01 |
| Bladder Urothelial Carcinoma | R217H | Missense_Mutation | Diploid | TCGA-2F-A9KQ-01 |
| Bladder Urothelial Carcinoma | S278R | Missense_Mutation | Gain | TCGA-4Z-AA80-01 |
| Bladder Urothelial Carcinoma | E396Q | Missense_Mutation | Diploid | TCGA-DK-A1AB-01 |
| Bladder Urothelial Carcinoma | R50H | Missense_Mutation | Diploid | TCGA-DK-A3WW-01 |
| Bladder Urothelial Carcinoma | S8L | Missense_Mutation | Gain | TCGA-XF-A9SJ-01 |
| Hepatocellular Carcinoma | Q81L | Missense_Mutation | Gain | TCGA-G3-A5SM-01 |
| Hepatocellular Carcinoma | S22I | Missense_Mutation | Diploid | TCGA-DD-AACY-01 |
| Hepatocellular Carcinoma | X209_splice | Splice_acceptor_variant | Gain | TCGA-CC-A8HV-01 |
| Hepatocellular Carcinoma | R424W | Missense_Mutation | Gain | TCGA-ED-A66Y-01 |
| Prostate Adenocarcinoma | T33M | Missense_Mutation | Diploid | TCGA-XK-AAIW-01 |
| Renal Clear Cell Carcinoma | L380F | Missense_Mutation | Diploid | TCGA-CJ-4869-01 |
| Endometrial Carcinoma *** | E221K | Missense_Mutation | Diploid | TCGA-A5-A0G2-01 |
| Uterine Endometrioid Carcinoma | R345H | Missense_Mutation | Diploid | TCGA-AJ-A3EL-01 |
| Uterine Endometrioid Carcinoma | D257N | Missense_Mutation | Diploid | TCGA-AP-A056-01 |
| Uterine Endometrioid Carcinoma | T266M | Missense_Mutation | Diploid | TCGA-AP-A059-01 |
| Uterine Endometrioid Carcinoma | E198Rfs*38 | Frame_Shift_Insertion | Diploid | TCGA-B5-A0JZ-01 |
| Uterine Endometrioid Carcinoma | R217C | Missense_Mutation | Diploid | TCGA-B5-A11E-01 |
| Uterine Endometrioid Carcinoma | E354K | Missense_Mutation | Gain | TCGA-BG-A0LX-01 |
| Uterine Endometrioid Carcinoma | L215V | Missense_Mutation | Diploid | TCGA-BG-A18C-01 |
| Uterine Endometrioid Carcinoma | E457K | Missense_Mutation | Diploid | TCGA-BS-A0TC-01 |
| Uterine Endometrioid Carcinoma | E349D | Missense_Mutation | Diploid | TCGA-EO-A22R-01 |
| Endometrial Carcinoma *** | S438 * | Nonsense_Mutation | Shallow Deletion | TCGA-EY-A1GS-01 |
| Endometrial Carcinoma *** | L380I | Missense_Mutation | Diploid | TCGA-A5-A0G2-01 |
| Uterine Endometrioid Carcinoma | E346 * | Nonsense_Mutation | Diploid | TCGA-A5-A2K5-01 |
| Uterine Endometrioid Carcinoma | R310S | Missense_Mutation | Diploid | TCGA-AJ-A3EK-01 |
| Uterine Endometrioid Carcinoma | R410= | Splice_Region, silent | Diploid | TCGA-AP-A1DV-01 |
| Uterine Endometrioid Carcinoma | L149F | Missense_Mutation | Diploid | TCGA-B5-A3F9-01 |
| Uterine Endometrioid Carcinoma | S325P | Missense_Mutation | Diploid | TCGA-B5-A3FC-01 |
| Uterine Endometrioid Carcinoma | V224L | Missense_Mutation | Diploid | TCGA-EO-A3AY-01 |
| Uterine Endometrioid Carcinoma | L131R | Missense_Mutation | Diploid | TCGA-EY-A1GD-01 |
| Uterine Endometrioid Carcinoma | F15V | Missense_Mutation | Diploid | TCGA-FI-A2D5-01 |
| Uterine Endometrioid Carcinoma | A247V | Missense_Mutation | Diploid | TCGA-FI-A2D5-01 |
| Lung Adenocarcinoma | E239K | Missense_Mutation | Gain | TCGA-05-4432-01 |
| Lung Adenocarcinoma | E225 * | Nonsense_Mutation | Gain | TCGA-44-7671-01 |
| Lung Adenocarcinoma | D259Y | Missense_Mutation | Gain | TCGA-73-A9RS-01 |
| Lung Adenocarcinoma | L234 * | Frame_Shift_Del | Gain | TCGA-86-A4JF-01 |
| Lung Adenocarcinoma | Q314 * | Nonsense_Mutation | Shallow Deletion | TCGA-95-7043-01 |
| Lung Adenocarcinoma | X209_splice | splice_acceptor_variant | Gain | TCGA-55-8301-01 |
| Esophagogastric Adenocarcinoma | K390T | Missense_Mutation | Shallow Deletion | TCGA-L5-A4OT-01 |
| Esophagogastric Adenocarcinoma | E288D | Missense_Mutation | Gain | TCGA-R6-A6DN-01 |
| Cutaneous Melanoma | S325F | Missense_Mutation | ShallowDel | TCGA-EE-A2GC-06 |
| Cutaneous Melanoma | P57R | Missense_Mutation | ShallowDel | TCGA-EE-A3JI-06 |
| Cutaneous Melanoma | P432L | Missense_Mutation | ShallowDel | TCGA-EE-A181-06 |
| Cutaneous Melanoma | E172K | Missense_Mutation | Diploid | TCGA-EB-A5UL-06 |
| Cutaneous Melanoma | L326F | Missense_Mutation | Not known | TCGA-EB-A41B-01 |
| Cutaneous Melanoma | S420F | Missense_Mutation | Diploid | TCGA-WE-A8K5-06 |
| Cutaneous Melanoma | R186L | Missense_Mutation | Diploid | TCGA-D3-A2JB-06 |
| Cutaneous Melanoma | R36S | Missense_Mutation | Diploid | TCGA-D3-A2JH-06 |
| Cutaneous Melanoma | S339F | Missense_Mutation | Diploid | TCGA-D3-A8GL-06 |
| Cutaneous Melanoma | E230K | Missense_Mutation | Gain | TCGA-D3-A8GM-06 |
| Cutaneous Melanoma | A287T | Missense_Mutation | Diploid | TCGA-W3-A824-06 |
| Cutaneous Melanoma | R155Q | Missense_Mutation | Diploid | TCGA-WE-A8K5-06 |
| Diffuse Type Stomach Adenocarcinoma | R345C | Missense_Mutation | Amplification | TCGA-HU-A4GU-01 |
| Mucinous Stomach Adenocarcinoma | R345H | Missense_Mutation | Diploid | TCGA-CD-8529-01 |
| Intestinal Type Stomach Adenocarcinoma | T266M | Missense_Mutation | Diploid | TCGA-VQ-A91K-01 |
| Tubular Stomach Adenocarcinoma | E349D | Missense_Mutation | Diploid | TCGA-BR-8680-01 |
| Stomach Adenocarcinoma | V434A | Missense_Mutation | Diploid | TCGA-BR-4292-01 |
| Intestinal Type Stomach Adenocarcinoma | R270H | Missense_Mutation | Diploid | TCGA-BR-7851-01 |
| Stomach Adenocarcinoma | L421P | Missense_Mutation | Diploid | TCGA-BR-8372-01 |
| Stomach Adenocarcinoma | X337_splice | splice_acceptor_variant | Diploid | TCGA-BR-8487-01 |
| Stomach Adenocarcinoma | Y319C | Missense_Mutation | Diploid | TCGA-BR-A4QM-01 |
| Diffuse Type Stomach Adenocarcinoma | F295S | Missense_Mutation | Diploid | TCGA-CD-A489-01 |
| Tubular Stomach Adenocarcinoma | E407D | Missense_Mutation | Shallow Deletion | TCGA-D7-6528-01 |
| Diffuse Type Stomach Adenocarcinoma | K390T | Missense_Mutation | Diploid | TCGA-D7-A747-01 |
| Breast Invasive Ductal Carcinoma | R450T | Missense_Mutation | Diploid | TCGA-C8-A12T-01 |
| Breast Invasive Ductal Carcinoma | E221K | Missense_Mutation | Diploid | TCGA-AC-A23H-01 |
| Breast Invasive Ductal Carcinoma | Q195H | Missense_Mutation | Diploid | TCGA-A7-A26H-01 |
| Breast Invasive Ductal Carcinoma | V77Cfs*34 | Frame_Shift_Deletion | Diploid | TCGA-D8-A27V-01 |
| Rectal Adenocarcinoma | R345C | Missense_Mutation | Diploid | TCGA-AG-3892-01 |
| Mucinous Adenocarcinoma of the Colon and Rectum | R310C | Missense_Mutation | Diploid | TCGA-CA-6717-01 |
| Mucinous Adenocarcinoma of the Colon and Rectum | R71W | Missense_Mutation | Diploid | TCGA-AD-5900-01 |
| Papillary Renal Cell Carcinoma | A308G | Missense_Mutation | Diploid | TCGA-DW-7842-01 |
| Head and Neck Squamous Cell Carcinoma | P57L | Missense_Mutation | Diploid | TCGA-CN-A640-01 |
| Head and Neck Squamous Cell Carcinoma | R304Q | Missense_Mutation | Amplification | TCGA-UF-A71D-01 |
| Head and Neck Squamous Cell Carcinoma | R304Q | Missense_Mutation | Gain | TCGA-CN-4727-01 |
| Head and Neck Squamous Cell Carcinoma | D211H | Missense_Mutation | Diploid | TCGA-CR-6481-01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.-T.; Waseem, A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers 2021, 13, 4985. https://doi.org/10.3390/cancers13194985
Usman S, Waseem NH, Nguyen TKN, Mohsin S, Jamal A, Teh M-T, Waseem A. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers. 2021; 13(19):4985. https://doi.org/10.3390/cancers13194985
Chicago/Turabian StyleUsman, Saima, Naushin H. Waseem, Thuan Khanh Ngoc Nguyen, Sahar Mohsin, Ahmad Jamal, Muy-Teck Teh, and Ahmad Waseem. 2021. "Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis" Cancers 13, no. 19: 4985. https://doi.org/10.3390/cancers13194985