
Senescence in HBV-, HCV- and NAFLD- Mediated Hepatocellular Carcinoma and Senotherapeutics: Current Evidence and Future Perspective

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Liver Cell Senescence: Definition and Characteristics
2.1. Definition
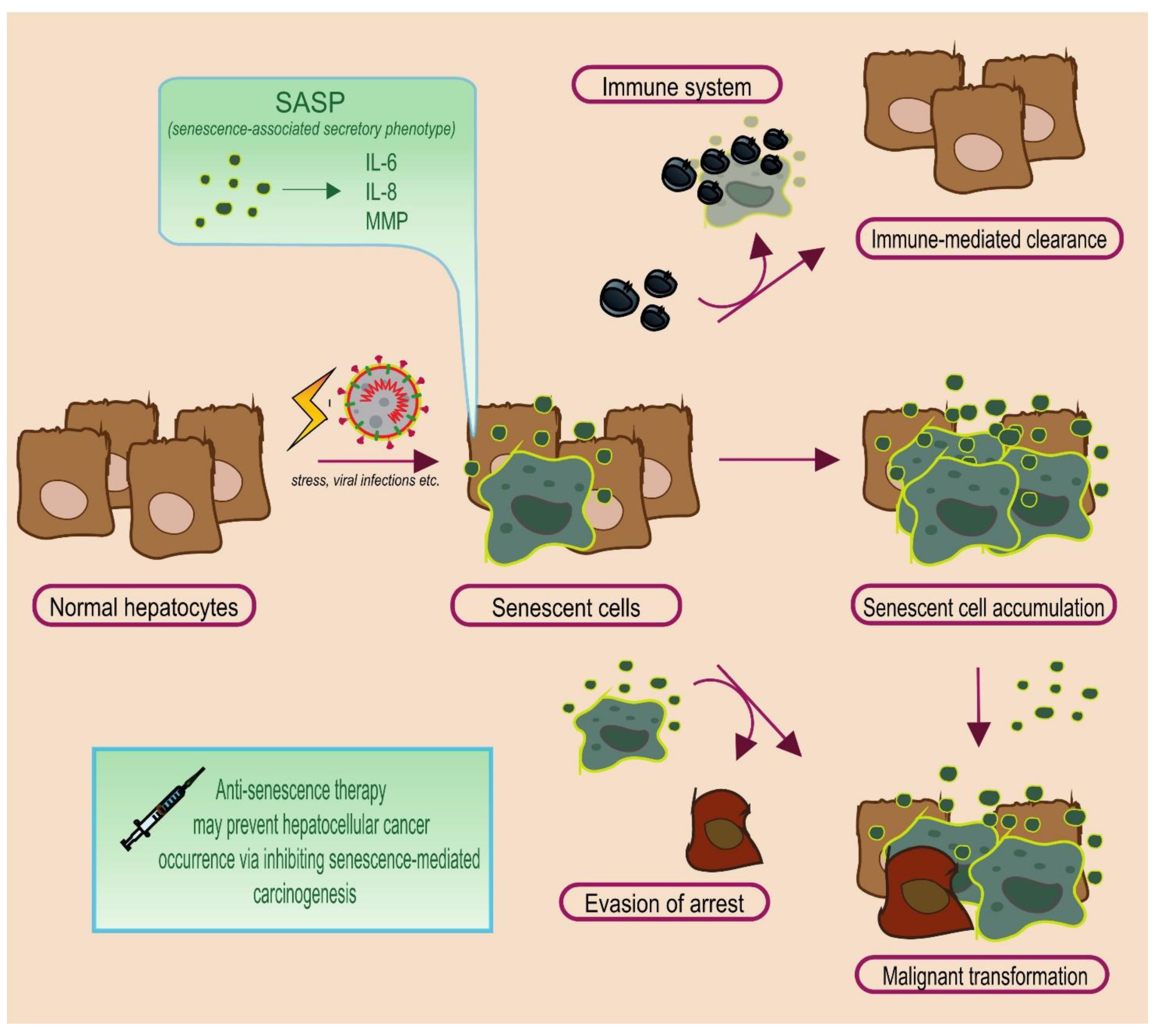
2.2. The Janus Face of Senescence: Senescence-Associated Secretory Phenotype
2.3. The Fate of Senescent Cells
3. Senescence in HBV-, HCV-, and NAFLD-Mediated HCC
3.1. Hepatitis B Virus (HBV): HBx Protein
3.2. Hepatitis C Virus (HCV): T-Cell Senescence and HCV Core Protein
3.3. Non-Alcoholic Fatty Liver Disease (NAFLD)
3.4. cGAS-STING Pathway: Linking Stress and Inflammation with Senescence and Cancer
4. Senotherapeutics in HCC: A Promising Field
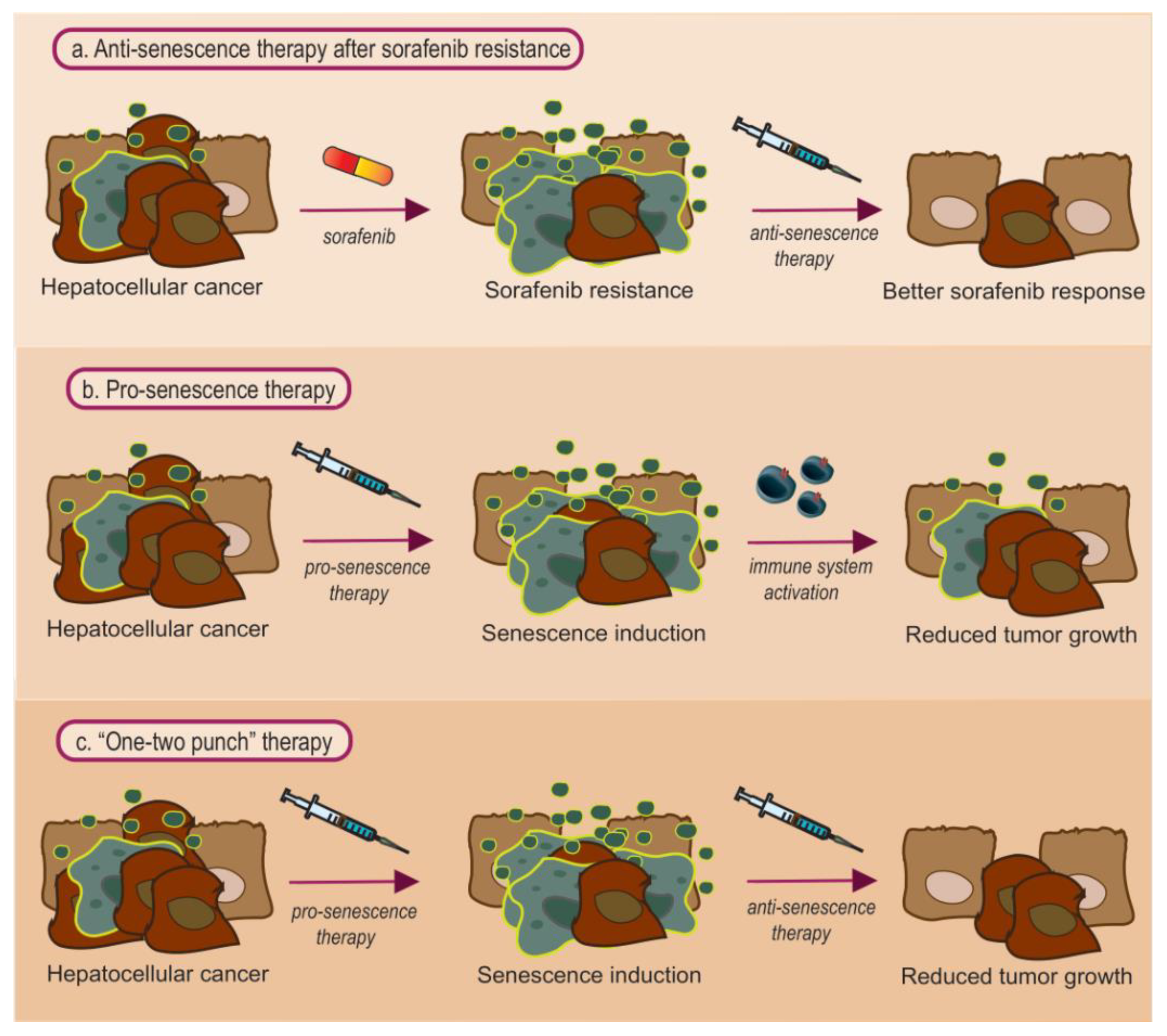
4.1. Anti-Senescence Therapy: Focus on Sorafenib Resistance
4.2. Pro-Senescence Therapy
4.3. Combined Pro- and Anti- Senescence Therapy: Is It the Best Approach?
5. Future Perspective
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Rattan, S.; Hayflick, L. Cellular Ageing and Replicative Senescence; Springer: Berlin/Heidelberg, Germany, 2016; pp. 350–351. [Google Scholar]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Huda, N.; Liu, G.; Hong, H.; Yan, S.; Khambu, B.; Yin, X.M. Hepatic senescence, the good and the bad. World J. Gastroenterol. 2019, 25, 5069–5081. [Google Scholar] [CrossRef]
- Martin, N.; Ziegler, D.V.; Parent, R.; Bernard, D. Hepatic Stellate Cell Senescence in Liver Tumorigenesis. Hepatology 2020, 73, 853–855. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target Ther. 2020, 5, 1–13. [Google Scholar] [CrossRef]
- Vogel, A.; Martinelli, E.; Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.-C.; Neumann, U.; et al. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim. Biophys. Acta-Rev. Cancer 2017, 1868, 564–570. [Google Scholar] [CrossRef]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.; Meyer, T.; Nault, J.C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv238–iv255. [Google Scholar] [CrossRef] [PubMed]
- Cabral, L.K.D.; Tiribelli, C.; Sukowati, C.H.C. Sorafenib resistance in hepatocellular carcinoma: The relevance of genetic heterogeneity. Cancers 2020, 12, 1576. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target Ther. 2020, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging (Albany N. Y.) 2016, 8, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Mittermeier, C.; Konopa, A.; Muehlich, S. Molecular Mechanisms to Target Cellular Senescence in Hepatocellular Carcinoma. Cells 2020, 9, 2540. [Google Scholar] [CrossRef]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.Y.; Kim, D.H.; Lee, E.K.; Chung, K.W.; Chung, S.; Lee, B.; Seo, A.Y.; Chung, J.H.; Jung, Y.S.; Im, E.; et al. Redefining chronic inflammation in aging and age-related diseases: Proposal of the senoinflammation concept. Aging Dis. 2019, 10, 367–382. [Google Scholar] [CrossRef] [Green Version]
- Tachtatzis, P.M.; Marshall, A.; Aravinthan, A.; Verma, S.; Penrhyn-Lowe, S.; Mela, M.; Scarpini, C.; Davies, S.E.; Coleman, N.; Alexander, G.J.M. Chronic hepatitis B virus infection: The relation between hepatitis B antigen expression, telomere length, senescence, inflammation and fibrosis. PLoS ONE 2015, 10, e0127511. [Google Scholar] [CrossRef] [Green Version]
- Papatheodoridi, A.M.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Nonalcoholic Fatty Liver Disease and Progression to Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 363–374. [Google Scholar] [CrossRef]
- Wandrer, F.; Han, B.; Liebig, S.; Schlue, J.; Manns, M.P.; Schulze-Osthoff, K.; Bantel, H. Senescence mirrors the extent of liver fibrosis in chronic hepatitis C virus infection. Aliment. Pharmacol. Ther. 2018, 48, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Rey, S.; Quintavalle, C.; Burmeister, K.; Calabrese, D.; Schlageter, M.; Quagliata, L.; Cathomas, G.; Diebold, J.; Molinolo, A.; Heim, M.H.; et al. Liver damage and senescence increases in patients developing hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2017, 32, 1480–1486. [Google Scholar] [CrossRef]
- Verma, S.; Tachtatzis, P.; Penrhyn-Lowe, S.; Scarpini, C.; Jurk, D.; Von Zglinicki, T.; Coleman, N.; Alexander, G.J.M. Sustained telomere length in hepatocytes and cholangiocytes with increasing age in normal liver. Hepatology 2012, 56, 1510–1520. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Idda, M.L.; Mcclusky, W.G.; Lodde, V.; Munk, R.; Abdelmohsen, K.; Rossi, M.; Gorospe, M. Survey of senescent cell markers with age in human tissues. Aging (Albany N. Y.) 2020, 12, 4052–4066. [Google Scholar] [CrossRef] [PubMed]
- Kovacovicova, K.; Skolnaja, M.; Heinmaa, M.; Mistrik, M.; Pata, P.; Pata, I.; Bartek, J.; Vinciguerra, M. Senolytic cocktail dasatinib+quercetin (D+Q) does not enhance the efficacy of senescence-inducing chemotherapy in liver cancer. Front. Oncol. 2018, 8, 459. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.S. Cellular senescence: A promising strategy for cancer therapy. BMB Rep. 2019, 52, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Brondello, J.M.; Prieur, A.; Philipot, D.; Lemaitre, J.M.; Lenaers, G.; Piette, J.; Dulić, V. La sénescence cellulaire: Un nouveau mythe de Janus? Med. Sci. 2012, 28, 288–296. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Miyazoe, Y.; Miuma, S.; Miyaaki, H.; Kanda, Y.; Nakashiki, S.; Sasaki, R.; Haraguchi, M.; Shibata, H.; Honda, T.; Taura, N.; et al. Extracellular vesicles from senescent hepatic stellate cells promote cell viability of hepatoma cells through increasing egf secretion from differentiated THP-1 cells. Biomed. Rep. 2020, 12, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Tang, Q.; Chen, M.; Chen, W.; Lu, Y.; Liu, Z.; He, Z. Hepatocellular Senescence: Immunosurveillance and Future Senescence-Induced Therapy in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 589908. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Chen, F.; Li, J.X.; Liu, C.C.; Zhang, H.B.; Xia, Y.; Yu, B.; You, P.; Xiang, D.; Lu, L.; et al. Reversal of hepatocyte senescence after continuous in vivo cell proliferation. Hepatology 2014, 60, 349–361. [Google Scholar] [CrossRef]
- Liu, L.; Yannam, G.R.; Nishikawa, T.; Yamamoto, T.; Basma, H.; Ito, R.; Nagaya, M.; Dutta-Moscato, J.; Stolz, D.B.; Duan, F.; et al. The microenvironment in hepatocyte regeneration and function in rats with advanced cirrhosis. Hepatology 2012, 55, 1529–1539. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P. What has senescence got to do with cancer? Cancer Cell 2005, 7, 505–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Koh, S.S.Y.; Lee, C.G.L. Hepatitis B virus X protein and hepatocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 940. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Jia, Z.; Tian, Y.; Yang, P.; Sun, H.; Wang, C.; Ding, Y.; Zhang, M.; Zhang, Y.; Yang, D.; et al. HBx Protein Contributes to Liver Carcinogenesis by H3K4me3 Modification Through Stabilizing WD Repeat Domain 5 Protein. Hepatology 2020, 71, 1678–1695. [Google Scholar] [CrossRef]
- Karakousis, N.D.; Papatheodoridi, A.; Chatzigeorgiou, A.; Papatheodoridis, G. Cellular senescence and hepatitis b related hepatocellular carcinoma: An intriguing link. Liver Int. 2020, 40, 2917–2927. [Google Scholar] [CrossRef]
- Toh, S.T.; Jin, Y.; Liu, L.; Wang, J.; Babrzadeh, F.; Gharizadeh, B.; Ronaghi, M.; Toh, H.C.; Chow, P.K.-H.; Chung, A.Y.-F.; et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis 2013, 34, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sze, K.M.F.; Chu, G.K.Y.; Lee, J.M.F.; Ng, I.O.L. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by c-jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013, 57, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, Q.; Gong, L.; Xu, H.; Liu, B.; Fang, X.; Yu, D.; Li, L.; Wei, T.; Wang, Y.; et al. C-terminal truncated HBx initiates hepatocarcinogenesis by downregulating TXNIP and reprogramming glucose metabolism. Oncogene 2021, 40, 1147–1161. [Google Scholar] [CrossRef]
- Ng, K.Y.; Chai, S.; Tong, M.; Guan, X.Y.; Lin, C.H.; Ching, Y.P.; Xie, D.; Sze-Lok, A.; Ma, S. C-terminal truncated hepatitis B virus X protein promotes hepatocellular carcinogenesis through induction of cancer and stem cell-like properties. Oncotarget 2016, 7, 24005–24017. [Google Scholar] [CrossRef]
- Idrissi, M.E.; Hachem, H.; Koering, C.; Merle, P.; Thénoz, M.; Mortreux, F.; Wattel, E. HBx triggers either cellular senescence or cell proliferation depending on cellular phenotype. J. Viral Hepat. 2016, 23, 130–138. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jung, J.K.; Lee, S.Y.; Jang, K.L. Hepatitis B virus X protein overcomes stress-induced premature senescence by repressing p16INK4a expression via DNA methylation. Cancer Lett. 2010, 288, 226–235. [Google Scholar] [CrossRef]
- Park, S.H.; Jung, J.K.; Lim, J.S.; Tiwari, I.; Jang, K.L. Hepatitis B virus X protein overcomes all-trans retinoic acid-induced cellular senescence by downregulating levels of p16 and p21 via DNA methylation. J. Gen. Virol. 2011, 92, 1309–1317. [Google Scholar] [CrossRef]
- Ou, D.P.; Tao, Y.M.; Tang, F.Q.; Yang, L.Y. The hepatitis B virus X protein promotes hepatocellular carcinoma metastasis by upregulation of matrix metalloproteinases. Int. J. Cancer 2007, 120, 1208–1214. [Google Scholar] [CrossRef]
- Zhou, X.; Yang, F.; Yang, Y.; Hu, Y.; Liu, W.; Huang, C.; Li, S.; Chen, Z. Cellular Physiology and Biochemistry Cellular Physiology and Biochemistry HBV Facilitated Hepatocellular Carcinoma Cells Proliferation by Up-Regulating Angiogenin Expression Through IL-6. Cell Physiol. Biochem. 2018, 46, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Sheng, T.; Wang, B.; Wang, S.Y.; Deng, B.; Qu, L.; Qi, X.S.; Wang, X.L.; Deng, G.L.; Sun, X. The Relationship Between Serum Interleukin-6 and the Recurrence of Hepatitis B Virus Related Hepatocellular Carcinoma after Curative Resection. Medicine 2015, 94, e941. [Google Scholar] [CrossRef]
- Su, J.M.; Lai, X.M.; Lan, K.H.; Li, C.P.; Chao, Y.; Yen, S.H.; Chang, F.Y.; Lee, S.D.; Lee, W.P. X protein of hepatitis B virus functions as a transcriptional corepressor on the human telomerase promoter. Hepatology 2007, 46, 402–413. [Google Scholar] [CrossRef]
- Wiemann, S.U.; Satyanarayana, A.; Tsahuridu, M.; Tillmann, H.L.; Zender, L.; Klempnauer, J.; Flemming, P.; Franco, S.; Blasco, M.A.; Manns, M.P.; et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J. 2002, 16, 935–942. [Google Scholar] [CrossRef]
- Paradis, V.; Youssef, N.; Dargère, D.; Bâ, N.; Bonvoust, F.; Deschatrette, J.; Bedossa, P. Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum. Pathol. 2001, 32, 327–332. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Sandhu, S.; Lai, J.P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Voehringer, D.; Blaser, C.; Brawand, P.; Raulet, D.H.; Hanke, T.; Pircher, H. Viral Infections Induce Abundant Numbers of Senescent CD8 T Cells. J. Immunol. 2001, 167, 4838–4843. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.S.; Park, S.H.; Jang, K.L. Hepatitis C virus Core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett. 2012, 321, 154–161. [Google Scholar] [CrossRef]
- Farinati, F.; Cardin, R.; Bortolami, M.; Burra, P.; Russo, F.P.; Rugge, M.; Guido, M.; Sergio, A.; Naccarato, R. Hepatitis C virus: From oxygen free radicals to hepatocellular carcinoma. J. Viral Hepat. 2007, 14, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Lim, J.S.; Lim, S.Y.; Tiwari, I.; Jang, K.L. Hepatitis C virus Core protein stimulates cell growth by down-regulating p16 expression via DNA methylation. Cancer Lett. 2011, 310, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Nakashima, T.; Okada, Y.; Jo, M.; Nishikawa, T.; Mitsumoto, Y.; Katagishi, T.; Kimura, H.; Itoh, Y.; Kagawa, K.; et al. Nuclear size measurement is a simple method for the assessment of hepatocellular aging in non-alcoholic fatty liver disease: Comparison with telomere-specific quantitative FISH and p21 immunohistochemistry. Pathol. Int. 2010, 60, 175–183. [Google Scholar] [CrossRef]
- Ping, F.; Li, Z.Y.; Lv, K.; Zhou, M.C.; Dong, Y.X.; Sun, Q.; Li, Y.X. Deoxyribonucleic acid telomere length shortening can predict the incidence of non-alcoholic fatty liver disease in patients with type 2 diabetes mellitus. J. Diabetes Investig. 2017, 8, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut microbiota promotes obesity-associated liver cancer through pge2-mediated suppression of antitumor immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [Green Version]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kahn, S.E. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans Is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.T.; Kanno, K.; Pham, Q.T.; Kikuchi, Y.; Kakimoto, M.; Kobayashi, T.; Otani, Y.; Kishikawa, N.; Miyauchi, M.; Arihiro, K.; et al. Senescent hepatic stellate cells caused by deoxycholic acid modulates malignant behavior of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 3255–3268. [Google Scholar] [CrossRef]
- Saga, K.; Iwashita, Y.; Hidano, S.; Aso, Y.; Isaka, K.; Kido, Y.; Tada, K.; Takayama, H.; Masuda, T.; Hirashita, T.; et al. Secondary unconjugated bile acids induce hepatic stellate cell activation. Int. J. Mol. Sci. 2018, 19, 3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Huangyang, P.; Burrows, M.; Guo, K.; Riscal, R.; Godfrey, J.; Lee, K.E.; Lin, N.; Lee, P.; Blair, I.A.; et al. FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat. Cell Biol. 2020, 22, 728–739. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450. [Google Scholar] [CrossRef]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Hüser, N.; Meiser, P.; Bayerl, F.; et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Du, J.; Zhu, H.; Ling, Q. The role of cGAS-STING signalling in liver diseases. JHEP Rep. 2021, 3, 100324. [Google Scholar] [CrossRef]
- Lauterbach-Rivière, L.; Bergez, M.; Mönch, S.; Qu, B.; Riess, M.; Vondran, F.W.R.; Liese, J.; Hornung, V.; Urban, S.; König, R. Hepatitis B Virus DNA is a Substrate for the cGAS/STING Pathway but is not Sensed in Infected Hepatocytes. Viruses 2020, 12, 592. [Google Scholar] [CrossRef]
- Ding, Q.; Cao, X.; Lu, J.; Huang, B.; Liu, Y.-J.; Kato, N.; Shu, H.-B.; Zhong, J. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 2013, 59, 52–58. [Google Scholar] [CrossRef]
- Luo, X.; Li, H.; Ma, L.; Zhou, J.; Guo, X.; Woo, S.-L.; Pei, Y.; Knight, L.R.; Deveau, M.; Chen, Y.; et al. Expression of STING Is Increased in Liver Tissues From Patients with NAFLD and Promotes Macrophage-Mediated Hepatic Inflammation and Fibrosis in Mice. Gastroenterology 2018, 155, 1971–1984.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase is a Cytosolic DNA Sensor that Activates the Type-I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.T.; Cui, C.; Qing, L.; Wang, L.S.; He, T.Y.; Yan, F.; Liu, F.Q.; Shen, Y.H.; Hou, X.G.; Chen, L. Activation of the STING-IRF3 pathway promotes hepatocyte inflammation, apoptosis and induces metabolic disorders in nonalcoholic fatty liver disease. Metab.-Clin. Exp. 2018, 81, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620. [Google Scholar] [CrossRef] [Green Version]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061. [Google Scholar] [CrossRef]
- Takahashi, A.; Loo, T.M.; Okada, R.; Kamachi, F.; Watanabe, Y.; Wakita, M.; Watanabe, S.; Kawamoto, S.; Miyata, K.; Barber, G.N.; et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402. [Google Scholar] [CrossRef] [Green Version]
- Loo, T.M.; Miyata, K.; Tanaka, Y.; Takahashi, A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. 2020, 111, 304. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.R.; Nelson, P.S. Cellular senescence and cancer chemotherapy resistance. Drug Resist. Updat. 2012, 15, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends in Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef]
- Serrano, M.; Barzilai, N. Targeting senescence. Nat. Med. 2018, 24, 1092–1094. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Niu, L.L.; Li, M.Y.; Yang, S.L.; Hu, B.G.; Chong, C.C.; Chan, S.L.; Ren, J.; Chen, G.G.; Lai, P.B. ID1-induced p16/IL6 axis activation contributes to the resistant of hepatocellular carcinoma cells to sorafenib. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Leung, C.O.N.; Tong, M.; Chung, K.P.S.; Zhou, L.; Che, N.; Tang, K.H.; Ding, J.; Lau, E.Y.T.; Ng, I.O.L.; Ma, S.; et al. Overriding Adaptive Resistance to Sorafenib Through Combination Therapy with Src Homology 2 Domain–Containing Phosphatase 2 Blockade in Hepatocellular Carcinoma. Hepatology 2020, 72, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Peng, H.; Xie, Y. BET family protein degraders poised to join the senolytic arsenal. Signal Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Muir, A.J.; Naggie, S. Hepatitis C Virus Treatment: Is It Possible To Cure All Hepatitis C Virus Patients? Clin. Gastroenterol. Hepatol. 2015, 13, 2166–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Serag, H.B.; Kanwal, F.; Richardson, P.; Kramer, J. Risk of Hepatocellular Carcinoma after Sustained Virologic Response in Veterans with HCV-infection Running Head: HCC after SVR. Hepatology 2016, 64, 130. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.; Kaspi, A.; Domovitz, T.; Davidovich, A.; Lavi-Itzkovitz, A.; Meirson, T.; Holmes, J.A.; Dai, C.-Y.; Huang, C.-F.; Chung, R.T.; et al. Hepatitis C virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet. 2019, 15. [Google Scholar] [CrossRef]
- Wong, C.-M.; Lee, J.M.-F.; Ching, Y.-P.; Jin, D.-Y.; Ng, I.O. Genetic and Epigenetic Alterations of DLC-1 Gene in Hepatocellular Carcinoma | Cancer Research. Cancer Res. 2003, 63, 7646–7651. [Google Scholar] [PubMed]
- Muehlich, S.; Gudermann, T. Pro-senescence therapy for hepatocellular carcinoma. Aging (Albany N. Y.) 2013, 5, 639–640. [Google Scholar] [CrossRef] [Green Version]
- Pipes, G.C.T.; Creemers, E.E.; Olson, E.N. The myocardin family of transcriptional coactivators: Versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006, 20, 1545–1556. [Google Scholar] [CrossRef] [Green Version]
- Hampl, V.; Martin, C.; Aigner, A.; Hoebel, S.; Singer, S.; Frank, N.; Sarikas, A.; Ebert, O.; Prywes, R.; Gudermann, T.; et al. Depletion of the transcriptional coactivators megakaryoblastic leukaemia 1 and 2 abolishes hepatocellular carcinoma xenograft growth by inducing oncogene-induced senescence. EMBO Mol. Med. 2013, 5, 1367–1382. [Google Scholar] [CrossRef]
- Wang, C.; Vegna, S.; Jin, H.; Benedict, B.; Lieftink, C.; Ramirez, C.; de Oliveira, R.L.; Morris, B.; Gadiot, J.; Wang, W.; et al. Inducing and exploiting vulnerabilities for the treatment of liver cancer. Nature 2019, 574, 268–272. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Liver Condition | Key Elements |
|---|---|
| HBV infection | Isoforms of the hepatitis B virus X (HBx) protein Different effects on senescence depending on the isoform Promotes factors of the SASP phenotype May enhance telomere shortening, and thus trigger replicative senescence and cirrhosis |
| HCV infection | Increased presence of senescent T-cells Do not effectively clear pre-malignant hepatocytes HCV core protein May promote the evasion of normal stress-induced senescence, allowing damaged cells to proliferate |
| NAFLD | Gut microbiota and senescent hepatic stellate cells Gut microbiota secrete DCA and LTA, which enter the circulation and promote hepatic stellate cell senescence as well as the expression of factors of the SASP phenotype |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannakoulis, V.G.; Dubovan, P.; Papoutsi, E.; Kataki, A.; Koskinas, J. Senescence in HBV-, HCV- and NAFLD- Mediated Hepatocellular Carcinoma and Senotherapeutics: Current Evidence and Future Perspective. Cancers 2021, 13, 4732. https://doi.org/10.3390/cancers13184732
Giannakoulis VG, Dubovan P, Papoutsi E, Kataki A, Koskinas J. Senescence in HBV-, HCV- and NAFLD- Mediated Hepatocellular Carcinoma and Senotherapeutics: Current Evidence and Future Perspective. Cancers. 2021; 13(18):4732. https://doi.org/10.3390/cancers13184732
Chicago/Turabian StyleGiannakoulis, Vassilis G., Peter Dubovan, Eleni Papoutsi, Agapi Kataki, and John Koskinas. 2021. "Senescence in HBV-, HCV- and NAFLD- Mediated Hepatocellular Carcinoma and Senotherapeutics: Current Evidence and Future Perspective" Cancers 13, no. 18: 4732. https://doi.org/10.3390/cancers13184732