
MiR-137 Targets the 3′ Untranslated Region of MSH2: Potential Implications in Lynch Syndrome-Related Colorectal Cancer

 , ,
, ,  , , , and
, , , and 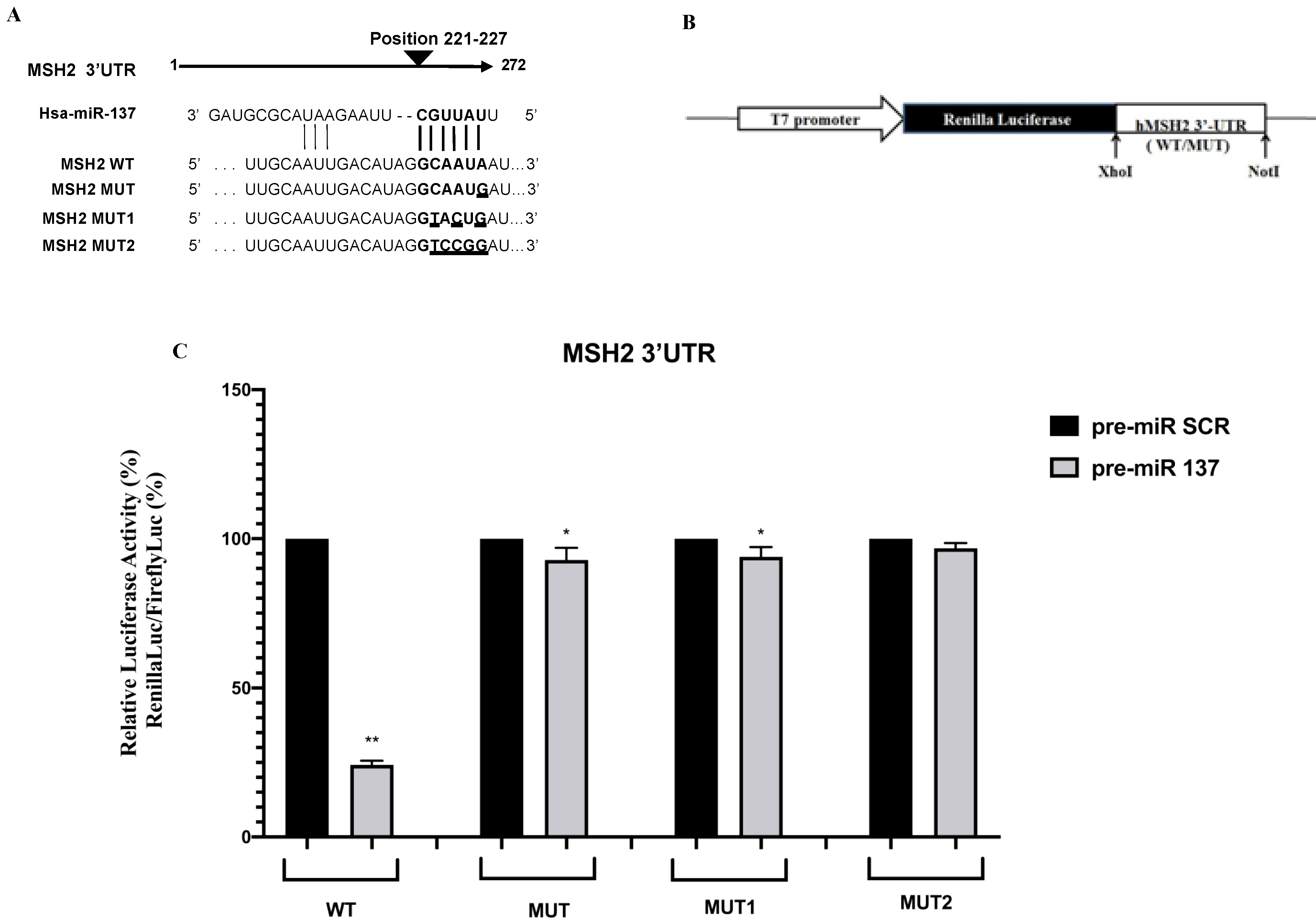{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Bioinformatic Analysis
2.2. Luciferase Constructs and Reporter Assay
2.3. Cell Culture and Transfection
2.4. SW480 Total RNA and Protein Analysis
2.5. Formalin-Fixed and Paraffin-Embedded (FFPE) Tissues RNA Isolation and Expression Assay
2.6. Analysis of Cell Proliferation
2.7. Statistical Analysis
3. Results
3.1. Identification of MiR-137 Using in Silico Analysis
3.2. Functional Effects of the MSH2 3′UTR Variant on Luciferase Reporter Expression
3.3. MiR-137 Regulates MSH2 Expression
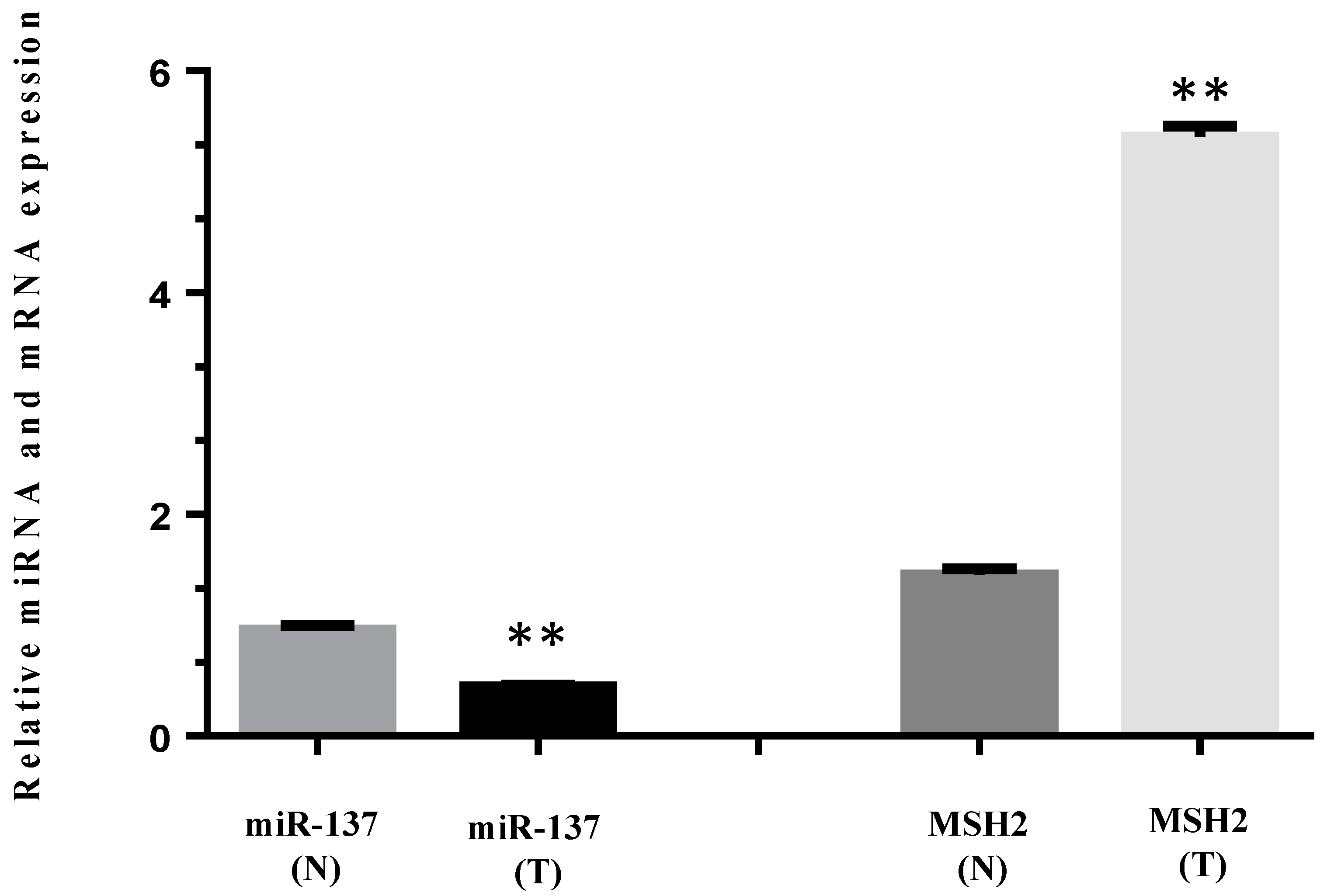
3.4. MiR-137 and mRNA MSH2 Levels Are Inversely Correlated in a Patient Carrying a 3′UTR Variant, the c.*226A>G
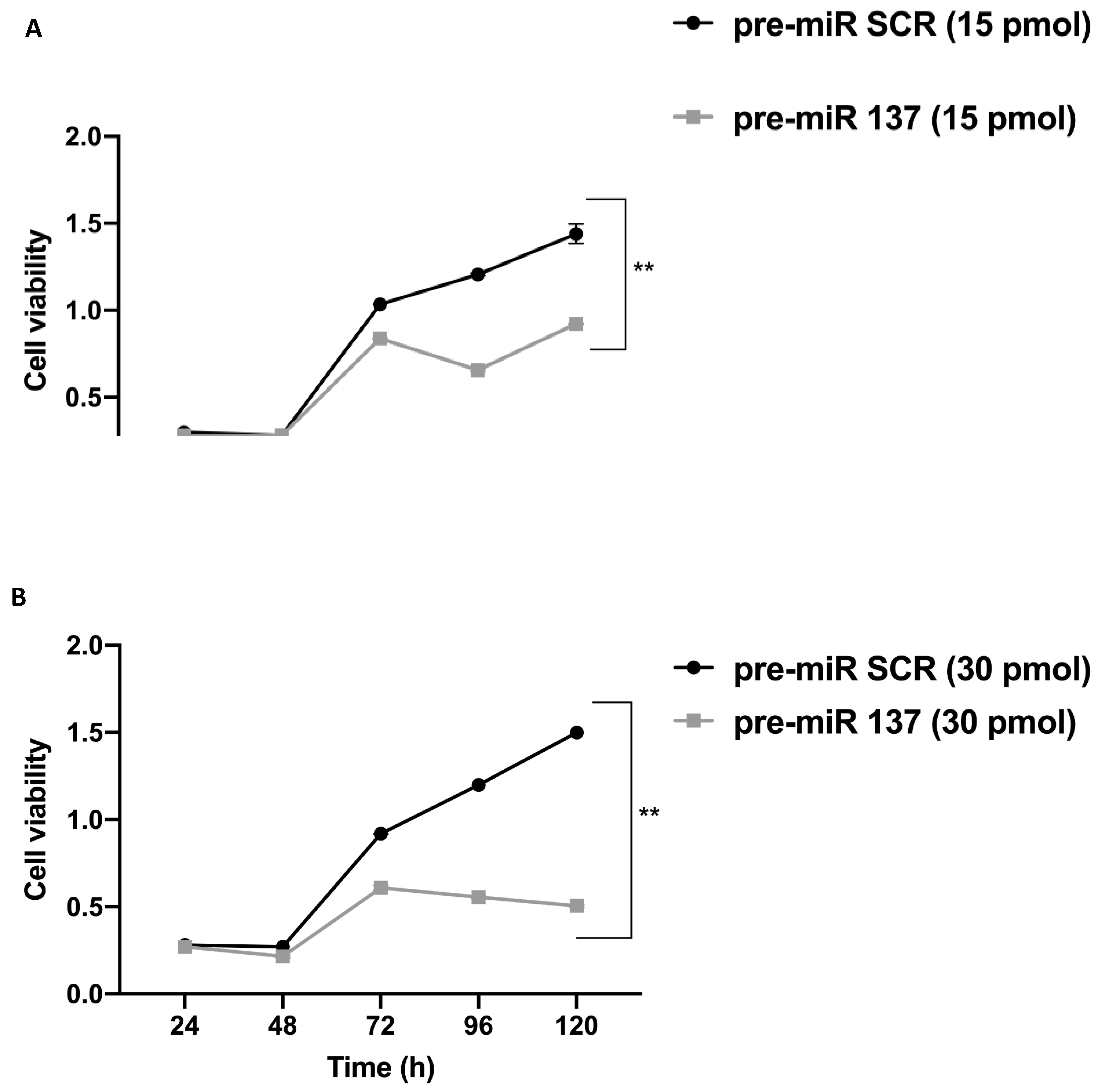
3.5. MiR-137 Effects on SW480 Cell Proliferation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pećina-Šlaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Duraturo, F.; Liccardo, R.; De Rosa, M.; Izzo, P. Genetics, diagnosis and treatment of Lynch syndrome: Old lessons and current challenges. Oncol. Lett. 2019, 17, 3048–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duraturo, F.; Liccardo, R.; Cavallo, A.; De Rosa, M.; Rossi, G.B.; Izzo, P. Multivariate analysis as a method for evaluating the pathogenicity of novel genetic MLH1 variants in patients with colorectal cancer and microsatellite instability. Int. J. Mol. Med. 2015, 36, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Duraturo, F.; Cavallo, A.; Liccardo, R.; Cudia, B.; De Rosa, M.; Diana, G.; Izzo, P. Contribution of large genomic rearrangements in Italian Lynch syndrome patients: Characterization of a novel alu-mediated deletion. Biomed. Res. Int. 2013, 2013, 219897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liccardo, R.; De Rosa, M.; Rossi, G.B.; Carlomagno, N.; Izzo, P.; Duraturo, F. Incomplete Segregation of MSH6 Frameshift Variants with Phenotype of Lynch Syndrome. Int. J. Mol. Sci. 2017, 18, 999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, R.M.; Gordhandas, S.; Maddy, B.P.; Baltich Nelson, B.; Askin, G.; Christos, P.J.; Caputo, T.A.; Chapman-Davis, E.; Holcomb, K.; Frey, M.K. Universal endometrial cancer tumor typing: How much has immunohistochemistry, microsatellite instability, and MLH1 methylation improved the diagnosis of Lynch syndrome across the population? Cancer 2019, 125, 3172–3183. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. The role of epigenetics in colorectal cancer. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 935–948. [Google Scholar] [CrossRef]
- Liccardo, R.; Nolano, A.; Lambiase, M.; Della Ragione, C.; De Rosa, M.; Izzo, P.; Duraturo, F. MSH2 Overexpression Due to an Unclassified Variant in 3′-Untranslated Region in a Patient with Colon Cancer. Biomedicines 2020, 8, 167. [Google Scholar] [CrossRef]
- Liccardo, R.; De Rosa, M.; Izzo, P.; Duraturo, F. Novel MSH2 splice-site mutation in a young patient with Lynch syndrome. Mol. Med. Rep. 2018, 17, 6942–6946. [Google Scholar] [CrossRef] [Green Version]
- Liccardo, R.; Della Ragione, C.; Mitilini, N.; De Rosa, M.; Izzo, P.; Duraturo, F. Novel variants of unknown significance in the PMS2 gene identified in patients with hereditary colon cancer. Cancer Manag. Res. 2019, 18, 6719–6725. [Google Scholar] [CrossRef] [Green Version]
- Liccardo, R.; De Rosa, M.; Rossi, G.B.; Rigler, G.; Izzo, P.; Duraturo, F. Characterization of novel, large duplications in the MSH2 gene of three unrelated Lynch syndrome patients. Cancer Genet. 2018, 221, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Duraturo, F.; Liccardo, R.; Izzo, P. Coexistence of MLH3 germline variants in colon cancer patients belonging to families with Lynch syndrome-associated brain tumors. J. Neurooncol. 2016, 129, 577–578. [Google Scholar] [CrossRef]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA mismatch repair deciency in sporadic colorectal cancer and Lynch syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Toft, N.J.; Winton, D.J.; Kelly, J.; Howard, L.A.; Dekker, M.; te Riele, H.; Arends, M.J.; Wyllie, A.H.; Margison, G.P.; Clarke, A.R. Msh2 status modulates both apoptosis and mutation frequency in the murine small intestine. Proc. Natl. Acad. Sci. USA 1999, 96, 3911–3915. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Richards, B.; Wilson, T.; Lloyd, M.; Cranston, A.; Thorburn, A.; Fishel, R.; Meuth, M. Apoptosis induced by overexpression of hMSH2 or hMLH1. Cancer Res. 1999, 59, 3021–3027. [Google Scholar]
- Tomlinson, I.; Bodmer, W. Selection, the mutation rate and cancer: Ensuring that the tail does not wag the dog. Nat. Med. 1999, 5, 11–12. [Google Scholar] [CrossRef]
- Stojic, L.; Mojas, N.; Cejka, P.; Di Pietro, M.; Ferrari, S.; Marra, G.; Jiricny, J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004, 18, 1331–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshioka, K.; Yoshioka, Y.; Hsieh, P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol. Cell 2006, 22, 501–510. [Google Scholar] [CrossRef]
- Mao, G.; Lee, S.; Ortega, J.; Gu, L.; Li, G.M. Modulation of microRNA processing by mismatch repair protein MutLα. Cell Res. 2012, 22, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Zhao, L.; Zhang, R.; Wei, Q.; Wang, M. Differential microRNA expression profiles associated with microsatellite status reveal possible epigenetic regulation of microsatellite instability in gastric adenocarcinoma. Ann. Transl. Med. 2020, 8, 484. [Google Scholar] [CrossRef] [PubMed]
- Svrcek, M.; El-Murr, N.; Wanherdrick, K.; Dumont, S.; Beaugerie, L.; Cosnes, J.; Colombel, J.F.; Tiret, E.; Fléjou, J.F.; Lesuffleur, T.; et al. Overexpression of microRNAs-155 and 21 targeting mismatch repair proteins in inflammatory bowel diseases. Carcinogenesis 2013, 34, 828–834. [Google Scholar] [CrossRef] [Green Version]
- Mao, G.; Pan, X.; Gu, L. Evidence that a mutation in the MLH1 3′-untranslated region confers a mutator phenotype and mismatch repair deficiency in patients with relapsed leukemia. J. Biol. Chem. 2008, 283, 3211–3216. [Google Scholar] [CrossRef] [Green Version]
- Cummins, J.M.; He, Y.; Leary, R.J.; Pagliarini, R.; Diaz, L.A., Jr.; Sjoblom, T.; Barad, O.; Bentwich, Z.; Szafranska, A.E.; Labourier, E.; et al. The colorectal microRNAome. Proc. Natl. Acad. Sci. USA 2006, 103, 3687–3692. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.A.; Frank, J.S. MicroRNAs in Mutagenesis, Genomic Instability and DNA Repair. Semin Oncol. 2011, 38, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Ann. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Yu, J.; Ng, S.S. MicroRNA dysregulation as a prognostic biomarker in colorectal cancer. Cancer Manag. Res. 2014, 6, 405–422. [Google Scholar]
- Bi, W.P.; Xia, M.; Wang, X.J. miR-137 suppresses proliferation, migration and invasion of colon cancer cell lines by targeting TCF4. Oncol. Lett. 2018, 15, 8744–8748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodaro, G.; Blasio, G.; Fiorentino, F.; Auberger, P.; Costanzo, P.; Cesaro, E. ZNF224 is a transcriptional repressor of AXL in chronic myeloid leukemia cells. Biochimie 2018, 154, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, E.; Pastore, A.; Polverino, A.; Manna, L.; Divisato, G.; Quintavalle, C.; Di Sanzo, M.; Faniello, M.C.; Grosso, M.; Costanzo, P. ZNF224 is a mediator of TGF-β pro-oncogenic function in melanoma. Hum. Mol. Genet. 2021, ddab173. [Google Scholar] [CrossRef]
- Montano, G.; Vidovic, K.; Palladino, C.; Cesaro, E.; Sodano, G.; Quintarelli, C.; De Angelis, B.; Errichiello, S.; Pane, F.; Izzo, P.; et al. WT1-mediated repression of the proapoptotic transcription factor ZNF224 is triggered by the BCR-ABL oncogene. Oncotarget 2015, 6, 28223–28237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turano, M.; Costabile, V.; Cerasuolo, A.; Duraturo, F.; Liccardo, R.; Delrio, P.; Pace, U.; Rega, D.; Dodaro, C.A.; Milone, M.; et al. Characterisation of mesenchymal colon tumour-derived cells in tumourspheres as a model for colorectal cancer progression. Int. J. Oncol. 2018, 53, 2379–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Li, J.; Li, J.; Wan, Y.; Li, T.; Ma, P.; Wang, Y.; Sang, H. Hsa-miR-137, hsa-miR-520e and hsa-miR-590-3p perform crucial roles in Lynch syndrome. Oncol. Lett. 2016, 12, 2011–2017. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Zeng, S. Relation between mismatch repair genes and colon cancer. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2014, 39, 190–194. [Google Scholar]
- Yamamoto, H.; Imai, K. Microsatellite instability: An update. Arch. Toxicol. 2015, 89, 899–921. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Adachi, Y.; Taniguchi, H.; Kunimoto, H.; Nosho, K.; Suzuki, H.; Shinomura, Y. Interrelationship between microsatellite instability and microRNA in gastrointestinal cancer. World J. Gastroenterol. 2012, 18, 2745–2755. [Google Scholar] [CrossRef]
- Smrt, R.D.; Szulwach, K.E.; Pfeiffer, R.L.; Li, X.; Guo, W.; Pathania, M.; Teng, Z.Q.; Luo, Y.; Peng, J.; Bordey, A.; et al. MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase Mind Bomb-1. Stem Cells. 2010, 28, 1060–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silber, J.; Lim, D.A.; Petritsch, C.; Persson, A.I.; Maunakea, A.K.; Yu, M.; Vandenberg, S.R.; Ginzinger, D.G.; James, C.D.; Costello, J.F.; et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008, 6, 14. [Google Scholar] [CrossRef]
- Liu, S.; Li, A.; Liu, Y.; Li, J.; Wang, M.; Sun, Y.; Qin, W.; Yu, C.; Jiang, T.; Liu, B. MIR137 polygenic risk is associated with schizophrenia and affects functional connectivity of the dorsolateral prefrontal cortex. Psychol. Med. 2020, 50, 1510–1518. [Google Scholar] [CrossRef]
- Jovčevska, I. Sequencing the next generation of glioblastomas. Crit. Rev. Clin. Lab. Sci. 2018, 55, 264–282. [Google Scholar] [CrossRef]
- Liu, M.; Lang, N.; Qiu, M.; Xu, F.; Li, Q.; Tang, Q.; Chen, J.; Chen, X.; Zhang, S.; Liu, Z.; et al. miR-137 targets Cdc42 expression, induces cell cycle G1 arrest and inhibits invasion in colorectal cancer cells. Int. J. Cancer 2010, 128, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, G.; Zhao, Y.; Han, Y.; Zhang, S.; Li, C.; Zhang, J. Long Noncoding RNA DSCAM-AS1 Facilitates Colorectal Cancer Cell Proliferation and Migration via miR-137/Notch1 Axis. J. Cancer 2020, 11, 6623–6632. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Yin, Y.; Peng, S.; Yang, G.; Yu, Y.; Guo, C.; Qin, Y.; Zhang, X.; Xu, W.; Qin, Y. Small nucleolar RNA host gene 1 promotes development and progression of colorectal cancer through negative regulation of miR-137. Mol. Carcinog. 2019, 58, 2104–2117. [Google Scholar] [CrossRef] [Green Version]
- Kashani, E.; Hadizadeh, M.; Chaleshi, V.; Mirfakhraie, R.; Young, C.; Savabkar, S.; Irani, S.; Asadzadeh Aghdaei, H.; Ashrafian Bonab, M. The Differential DNA Hypermethylation Patterns of microRNA-137 and microRNA-342 Locus in Early Colorectal Lesions and Tumours. Biomolecules 2019, 9, 519. [Google Scholar] [CrossRef] [Green Version]
- Bemis, L.T.; Chen, R.; Amato, C.M.; Classen, E.H.; Robinson, S.E.; Coffey, D.G.; Erickson, P.F.; Shellman, Y.G.; Robinson, W.A. MicroRNA-137 targets microphthalmia-associated transcription factor in melanoma cell lines. Cancer Res. 2008, 68, 1362–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langevin, S.M.; Stone, R.A.; Bunker, C.H.; Grandis, J.R.; Sobol, R.W.; Taioli, E. MicroRNA-137 promoter methylation in oral rinses from patients with squamous cell carcinoma of the head and neck is associated with gender and body mass index. Carcinogenesis 2010, 31, 864–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langevin, S.M.; Stone, R.A.; Bunker, C.H.; Lyons-Weiler, M.A.; LaFramboise, W.A.; Kelly, L.; Seethala, R.R.; Grandis, J.R.; Sobol, R.W.; Taioli, E. MicroRNA-137 promoter methylation is associated with poorer overall survival in patients with squamous cell carcinoma of the head and neck. Cancer 2011, 117, 1454–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaguer, F.; Link, A.; Lozano, J.J.; Cuatrecasas, M.; Nagasaka, T.; Boland, C.R.; Goel, A. Epigenetic silencing of miR-137 is an early event in colorectal carcinogenesis. Cancer Res. 2010, 70, 6609–6618. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, J.H.; Shan, T.; Aguilera-Barrantes, I.; Wang, L.S.; Huang, T.H.M.; Rader, J.S.; Sheng, X.; Huang, Y.W. miR-137 is a tumor suppressor in endometrial cancer and is repressed by DNA hypermethylation. Lab. Investig. 2018, 98, 1397–1407. [Google Scholar] [CrossRef] [Green Version]
- Banno, K.; Yanokura, M.; Iida, M.; Masuda, K.; Aoki, D. Carcinogenic mechanisms of endometrial cancer: Involvement of genetics and epigenetics. J. Obstet. Gynaecol. Res. 2014, 40, 1957–1967. [Google Scholar] [CrossRef]
- Hassen, S.; Ali, A.A.; Kilaparty, S.P.; Al-Anbaky, Q.A.; Majeed, W.; Boman, B.M.; Fields, J.Z.; Ali, N. Interdependence of DNA mismatch repair proteins MLH1 and MSH2 in apoptosis in human colorectal carcinoma cell lines. Mol. Cell. Biochem. 2016, 412, 297–305. [Google Scholar] [CrossRef]
- Mendez-Bermudez, A.; Royle, N.J. Deficiency in DNA mismatch repair increases the rate of telomere shortening in normal human cells. Hum. Mutat. 2011, 32, 939–946. [Google Scholar] [CrossRef]
- Cerretelli, G.; Ager, A.; Arends, M.J.; Frayling, I.M. Molecular pathology of Lynch syndrome. J. Pathol. 2020, 250, 518–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, D.; Lin, B.; Cowan, A.; Heinen, C.D. ATR-Chk1 activation mitigates replication stress caused by mismatch repair-dependent processing of DNA damage. Proc. Natl. Acad. Sci. USA 2018, 115, 1523–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turano, M.; Delrio, P.; Rega, D.; Cammarota, F.; Polverino, A.; Duraturo, F.; Izzo, P.; De Rosa, M. Promising Colorectal Cancer Biomarkers for Precision Prevention and Therapy. Cancers 2019, 11, 1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liccardo, R.; Sessa, R.; Trombetti, S.; De Rosa, M.; Izzo, P.; Grosso, M.; Duraturo, F. MiR-137 Targets the 3′ Untranslated Region of MSH2: Potential Implications in Lynch Syndrome-Related Colorectal Cancer. Cancers 2021, 13, 4662. https://doi.org/10.3390/cancers13184662
Liccardo R, Sessa R, Trombetti S, De Rosa M, Izzo P, Grosso M, Duraturo F. MiR-137 Targets the 3′ Untranslated Region of MSH2: Potential Implications in Lynch Syndrome-Related Colorectal Cancer. Cancers. 2021; 13(18):4662. https://doi.org/10.3390/cancers13184662
Chicago/Turabian StyleLiccardo, Raffaella, Raffaele Sessa, Silvia Trombetti, Marina De Rosa, Paola Izzo, Michela Grosso, and Francesca Duraturo. 2021. "MiR-137 Targets the 3′ Untranslated Region of MSH2: Potential Implications in Lynch Syndrome-Related Colorectal Cancer" Cancers 13, no. 18: 4662. https://doi.org/10.3390/cancers13184662