
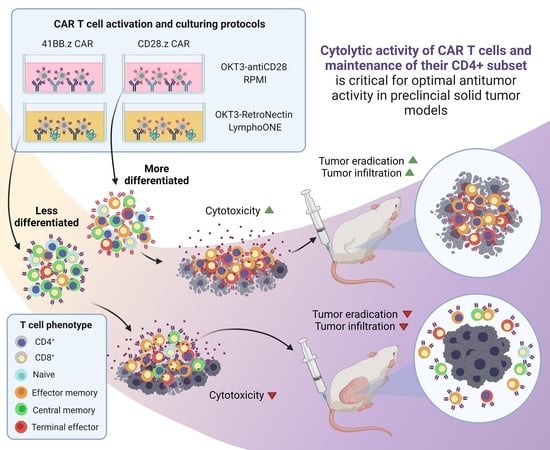
Cytolytic Activity of CAR T Cells and Maintenance of Their CD4+ Subset Is Critical for Optimal Antitumor Activity in Preclinical Solid Tumor Models

 , and
, and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Retrovirus Production and Transduction of T Cells
2.3. Flow Cytometry
2.4. In Vitro Rechallenge Assays
2.5. Cytokine Secretion Assay
2.6. Cytotoxicity Assay
2.7. Phospho-Proteome Array
2.8. Cluster Analysis
2.9. Xenograft Tumors and In Vivo Car T Cell Treatments
2.10. Statistical Analysis
3. Results
3.1. The OKT3-RetroNectin/LymphoONE CAR T Cell Generation Protocol Restrains T Cell Differentiation
3.2. The Effects of HER2-CAR T Cell Phenotype on Cytokine Production Post-Antigen-Specific Activation Depends on the Utilized Costimulatory Domain and the Method of CAR Activation
3.3. Differentiated CAR T Cell Products Induce Stronger In Vitro Cytotoxicity
3.4. Differentiated CAR T Cell Products Have a Greater Proliferative Capacity upon Repeated Stimuation In Vitro
3.5. Effector Memory Phenotype-Enriched Car T Cell Products Induce Complete Tumor Eradication In Vivo
3.6. Effector Memory Phenotype-Enriched Car T Cell Products Show Faster Expansion In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Reagan, P.M.; Friedberg, J.W. Axicabtagene ciloleucel and brexucabtagene autoleucel in relapsed and refractory diffuse large B-cell and mantle cell lymphomas. Future Oncol. 2021, 17, 1269–1283. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhao, P.; Huang, H. Engineering better chimeric antigen receptor T cells. Exp. Hematol. Oncol. 2020, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Abken, H. Costimulation Engages the Gear in Driving CARs. Immunity 2016, 44, 214–216. [Google Scholar] [CrossRef] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatino, M.; Hu, J.; Sommariva, M.; Gautam, S.; Fellowes, V.; Hocker, J.D.; Dougherty, S.; Qin, H.; Klebanoff, C.A.; Fry, T.J.; et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood 2016, 128, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Popplewell, L.L.; Wagner, J.R.; Naranjo, A.; Blanchard, M.S.; Mott, M.R.; Norris, A.P.; Wong, C.W.; Urak, R.Z.; Chang, W.C.; et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood 2016, 127, 2980–2990. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.S.; Nukaya, I.; Enoki, T.; Chatani, E.; Kato, A.; Goto, Y.; Dan, K.; Sasaki, M.; Tomita, K.; Tanabe, M.; et al. In vivo persistence of genetically modified T cells generated ex vivo using the fibronectin CH296 stimulation method. Cancer Gene Ther. 2008, 15, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Gargett, T.; Brown, M.P. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy 2015, 17, 487–495. [Google Scholar] [CrossRef]
- Stock, S.; Hoffmann, J.M.; Schubert, M.L.; Wang, L.; Wang, S.; Gong, W.; Neuber, B.; Gern, U.; Schmitt, A.; Muller-Tidow, C.; et al. Influence of Retronectin-Mediated T-Cell Activation on Expansion and Phenotype of CD19-Specific Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 1167–1182. [Google Scholar] [CrossRef]
- Hosoi, H.; Ikeda, H.; Imai, N.; Amaike, C.; Wang, L.; Orito, Y.; Yamane, M.; Ueno, H.; Ideno, M.; Nukaya, I.; et al. Stimulation through very late antigen-4 and -5 improves the multifunctionality and memory formation of CD8(+) T cells. Eur. J. Immunol. 2014, 44, 1747–1758. [Google Scholar] [CrossRef]
- Medvec, A.R.; Ecker, C.; Kong, H.; Winters, E.A.; Glover, J.; Varela-Rohena, A.; Riley, J.L. Improved Expansion and In Vivo Function of Patient T Cells by a Serum-free Medium. Mol. Ther. Methods Clin. Dev. 2018, 8, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Mata, M.; Gerken, C.; Nguyen, P.; Krenciute, G.; Spencer, D.M.; Gottschalk, S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov. 2017, 7, 1306–1319. [Google Scholar] [CrossRef] [Green Version]
- Tanner, M.; Kapanen, A.I.; Junttila, T.; Raheem, O.; Grenman, S.; Elo, J.; Elenius, K.; Isola, J. Characterization of a novel cell line established from a patient with Herceptin-resistant breast cancer. Mol. Cancer Ther. 2004, 3, 1585–1592. [Google Scholar]
- Ahmed, N.; Salsman, V.S.; Yvon, E.; Louis, C.U.; Perlaky, L.; Wels, W.S.; Dishop, M.K.; Kleinerman, E.E.; Pule, M.; Rooney, C.M.; et al. Immunotherapy for osteosarcoma: Genetic modification of T cells overcomes low levels of tumor antigen expression. Mol. Ther. 2009, 17, 1779–1787. [Google Scholar] [CrossRef]
- Cobbold, S.P.; Jayasuriya, A.; Nash, A.; Prospero, T.D.; Waldmann, H. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature 1984, 312, 548–551. [Google Scholar] [CrossRef]
- Qin, S.X.; Wise, M.; Cobbold, S.P.; Leong, L.; Kong, Y.C.; Parnes, J.R.; Waldmann, H. Induction of tolerance in peripheral T cells with monoclonal antibodies. Eur. J. Immunol. 1990, 20, 2737–2745. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Szoor, A.; Toth, G.; Zsebik, B.; Szabo, V.; Eshhar, Z.; Abken, H.; Vereb, G. Trastuzumab derived HER2-specific CARs for the treatment of trastuzumab-resistant breast cancer: CAR T cells penetrate and eradicate tumors that are not accessible to antibodies. Cancer Lett. 2020, 484, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef] [Green Version]
- Moller-Hackbarth, K.; Dewitz, C.; Schweigert, O.; Trad, A.; Garbers, C.; Rose-John, S.; Scheller, J. A disintegrin and metalloprotease (ADAM) 10 and ADAM17 are major sheddases of T cell immunoglobulin and mucin domain 3 (Tim-3). J. Biol. Chem. 2013, 288, 34529–34544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilber, E.; Martin, G.E.; Willberg, C.B.; Fox, J.; Nwokolo, N.; Fidler, S.; Frater, J.; Investigators, C. Soluble plasma programmed death 1 (PD-1) and Tim-3 in primary HIV infection. AIDS 2019, 33, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Jacinto, E.; Hibi, M.; Kallunki, T.; Karin, M.; Ben-Neriah, Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell 1994, 77, 727–736. [Google Scholar] [CrossRef]
- Colombetti, S.; Basso, V.; Mueller, D.L.; Mondino, A. Prolonged TCR/CD28 engagement drives IL-2-independent T cell clonal expansion through signaling mediated by the mammalian target of rapamycin. J. Immunol. 2006, 176, 2730–2738. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009, 15, 808–813. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, S.; Zhao, D.M.; Harty, J.T.; Badovinac, V.P.; Xue, H.H. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 2010, 33, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Ji, Y.; Gattinoni, L.; Zhang, L.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol. Immunother. 2013, 62, 727–736. [Google Scholar] [CrossRef]
- Refaeli, Y.; Van Parijs, L.; London, C.A.; Tschopp, J.; Abbas, A.K. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity 1998, 8, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Guan, X.Y.; Jiang, P. Cytokine and Chemokine Signals of T-Cell Exclusion in Tumors. Front. Immunol. 2020, 11, 594609. [Google Scholar] [CrossRef]
- Nie, Y.; Lu, W.; Chen, D.; Tu, H.; Guo, Z.; Zhou, X.; Li, M.; Tu, S.; Li, Y. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. Biomark Res. 2020, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Junghans, R.P. The challenges of solid tumor for designer CAR-T therapies: A 25-year perspective. Cancer Gene Ther. 2017, 24, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barok, M.; Isola, J.; Palyi-Krekk, Z.; Nagy, P.; Juhasz, I.; Vereb, G.; Kauraniemi, P.; Kapanen, A.; Tanner, M.; Vereb, G.; et al. Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance. Mol. Cancer Ther. 2007, 6, 2065–2072. [Google Scholar] [CrossRef] [Green Version]
- Palyi-Krekk, Z.; Barok, M.; Isola, J.; Tammi, M.; Szollosi, J.; Nagy, P. Hyaluronan-induced masking of ErbB2 and CD44-enhanced trastuzumab internalisation in trastuzumab resistant breast cancer. Eur. J. Cancer 2007, 43, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Toth, G.; Szollosi, J.; Abken, H.; Vereb, G.; Szoor, A. A Small Number of HER2 Redirected CAR T Cells Significantly Improves Immune Response of Adoptively Transferred Mouse Lymphocytes against Human Breast Cancer Xenografts. Int. J. Mol. Sci. 2020, 21, 1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 1–16. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Textor, A.; Grunewald, L.; Anders, K.; Klaus, A.; Schwiebert, S.; Winkler, A.; Stecklum, M.; Rolff, J.; Henssen, A.G.; Hopken, U.E.; et al. CD28 Co-Stimulus Achieves Superior CAR T Cell Effector Function against Solid Tumors Than 4-1BB Co-Stimulus. Cancers 2021, 13, 1050. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Rietberg, S.P.; Sotillo, E.; Dong, R.; Vachharajani, V.T.; Labanieh, L.; Myklebust, J.H.; Kadapakkam, M.; Weber, E.W.; Tousley, A.M.; et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020, 10, 702–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, M.; Gottschalk, S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front. Immunol. 2021, 12, 684642. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kohler, M.E.; Chien, C.D.; Sauter, C.T.; Jacoby, E.; Yan, C.; Hu, Y.; Wanhainen, K.; Qin, H.; Fry, T.J. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci. Transl. Med. 2017, 9, eaag1209. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Aguilar, B.; Starr, R.; Alizadeh, D.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; Forman, S.J.; Brown, C.E. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity. JCI Insight 2018, 3, e99048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, A.; Khan, I.; Kaplan, M. T helper differentiation of CAR T cells augments function and target cell killing. J. Immunol. 2020, 204 (Suppl. 1), 246.6. [Google Scholar]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Csaplár, M.; Szöllősi, J.; Gottschalk, S.; Vereb, G.; Szöőr, Á. Cytolytic Activity of CAR T Cells and Maintenance of Their CD4+ Subset Is Critical for Optimal Antitumor Activity in Preclinical Solid Tumor Models. Cancers 2021, 13, 4301. https://doi.org/10.3390/cancers13174301
Csaplár M, Szöllősi J, Gottschalk S, Vereb G, Szöőr Á. Cytolytic Activity of CAR T Cells and Maintenance of Their CD4+ Subset Is Critical for Optimal Antitumor Activity in Preclinical Solid Tumor Models. Cancers. 2021; 13(17):4301. https://doi.org/10.3390/cancers13174301
Chicago/Turabian StyleCsaplár, Marianna, János Szöllősi, Stephen Gottschalk, György Vereb, and Árpád Szöőr. 2021. "Cytolytic Activity of CAR T Cells and Maintenance of Their CD4+ Subset Is Critical for Optimal Antitumor Activity in Preclinical Solid Tumor Models" Cancers 13, no. 17: 4301. https://doi.org/10.3390/cancers13174301