
The Role of Chemotherapy in Management of Inoperable, Metastatic and/or Recurrent Melanotic Neuroectodermal Tumor of Infancy—Own Experience and Systematic Review

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
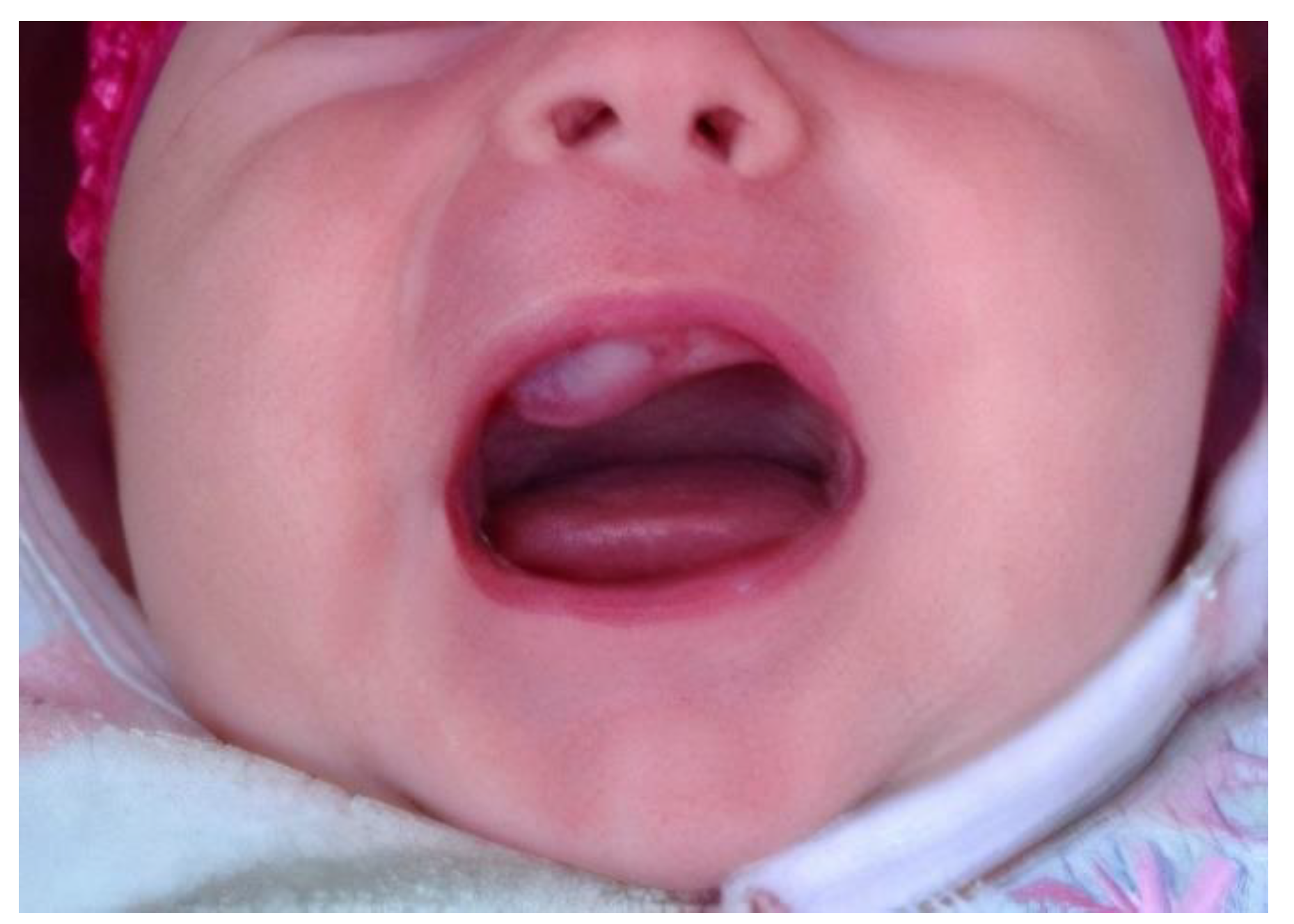
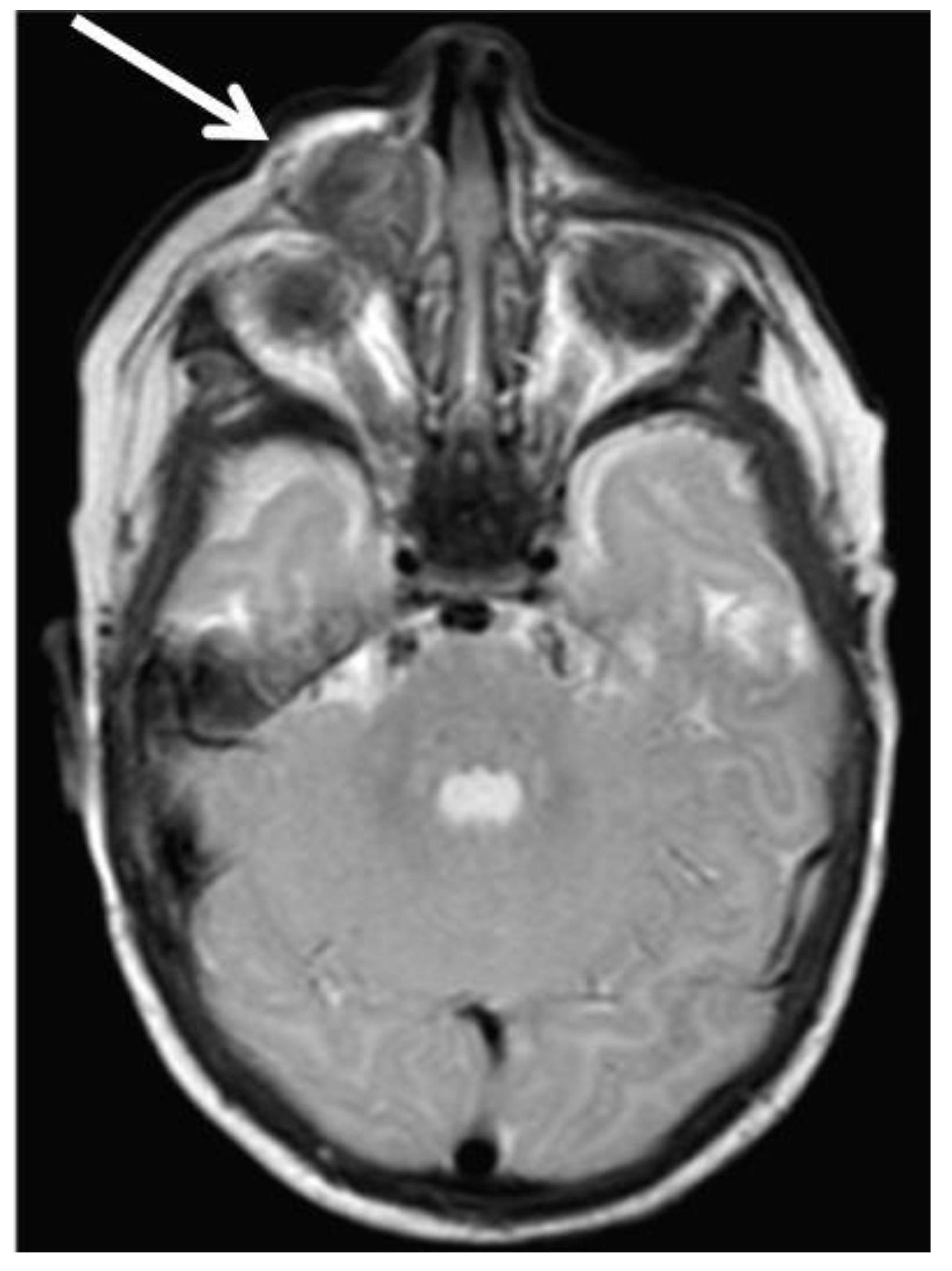
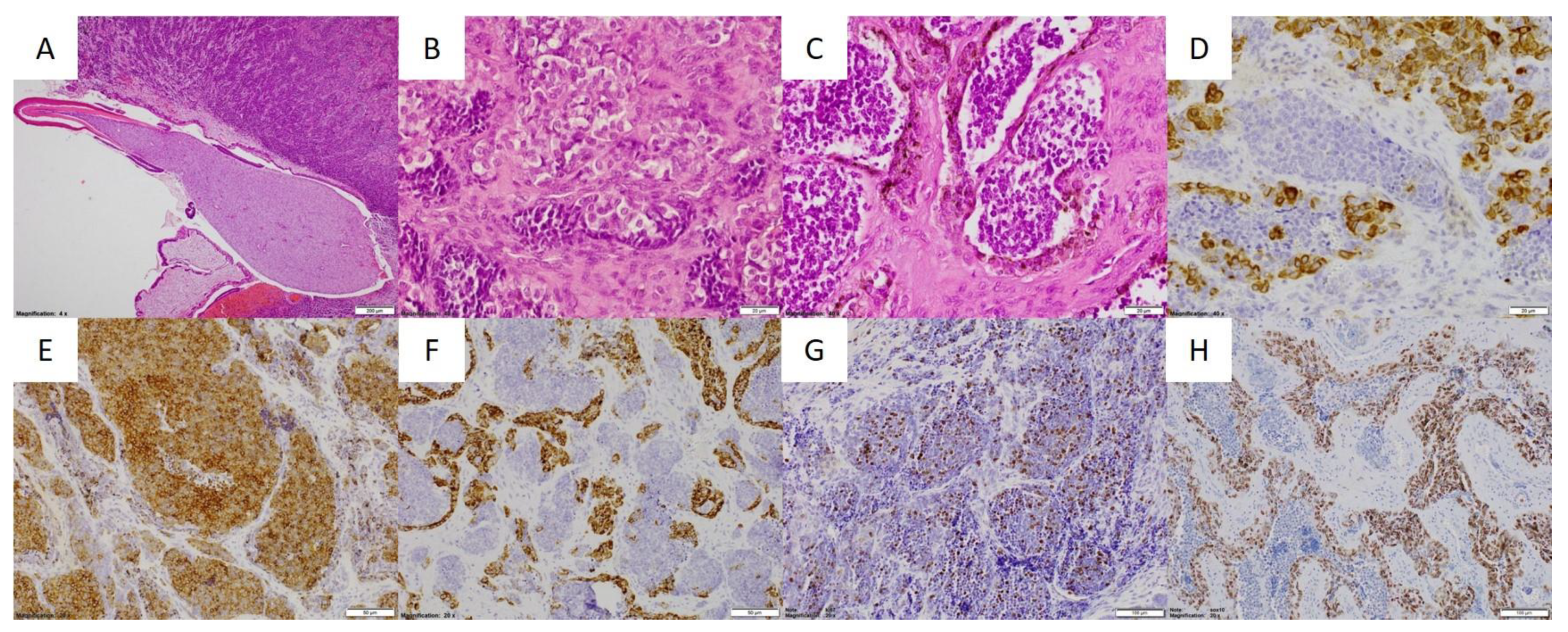
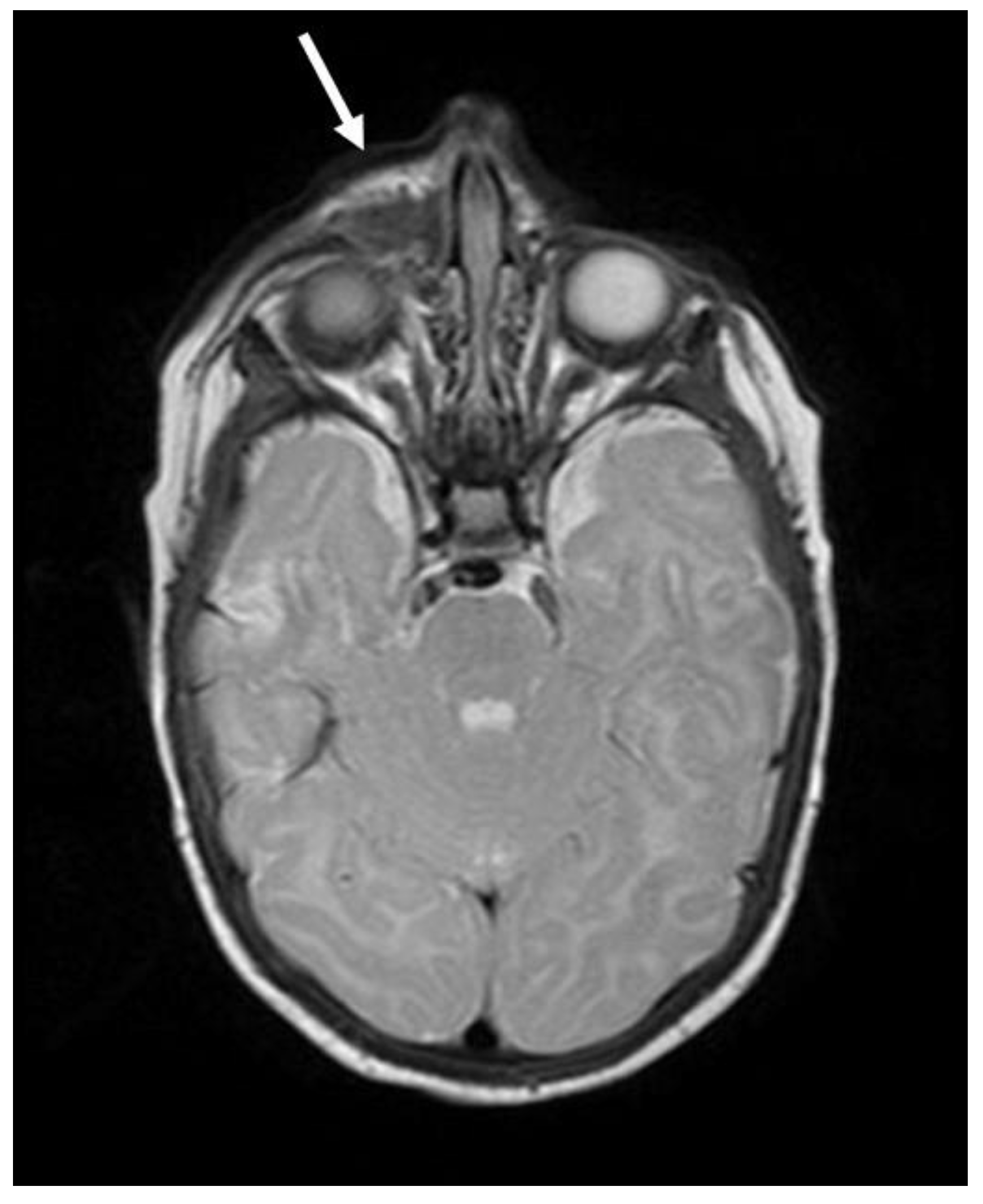
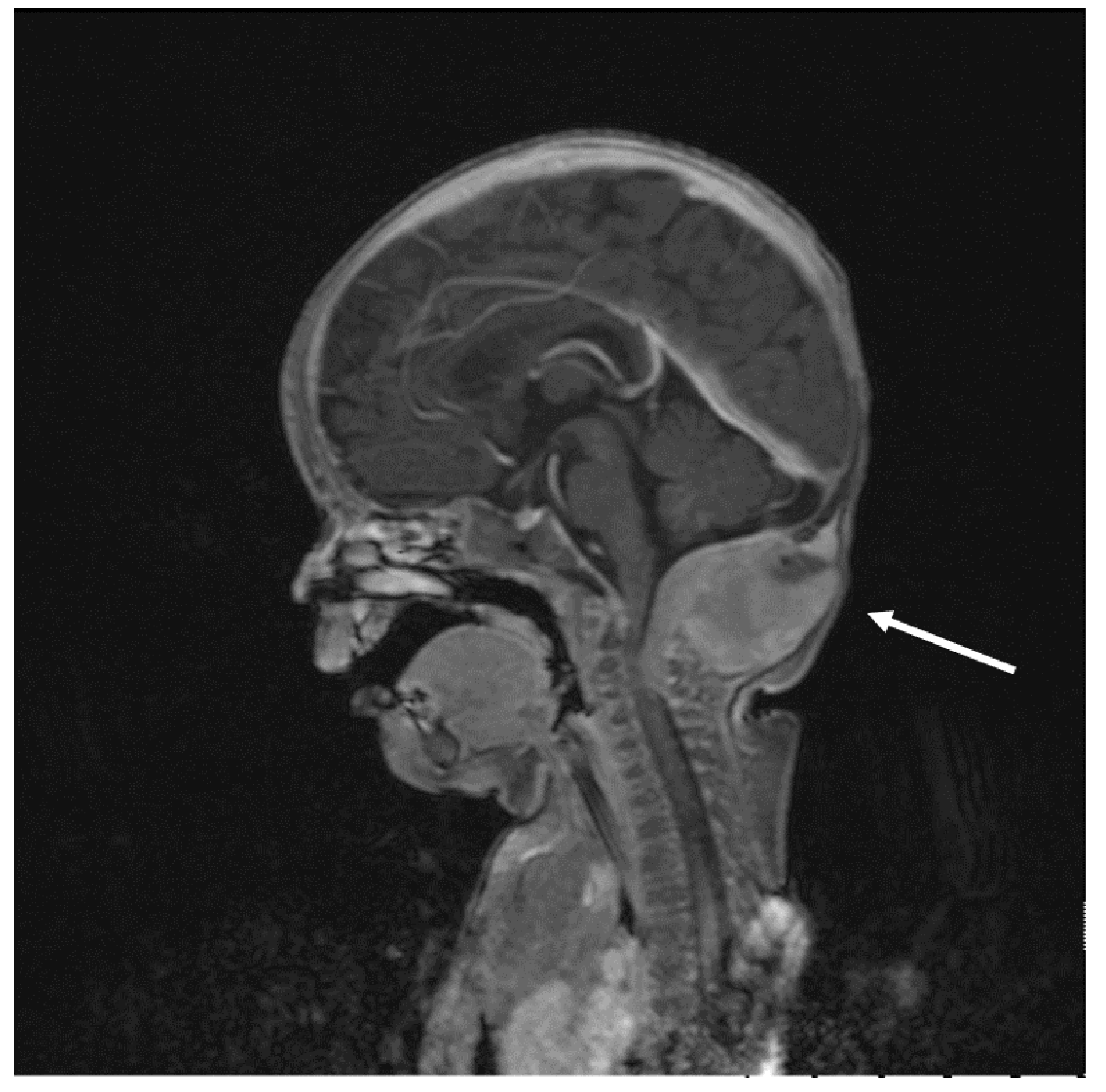
2. Case Reports
2.1. Patient 1
2.2. Patient 2
2.3. Patient 3
2.4. Patient 4
3. Review of the Literature
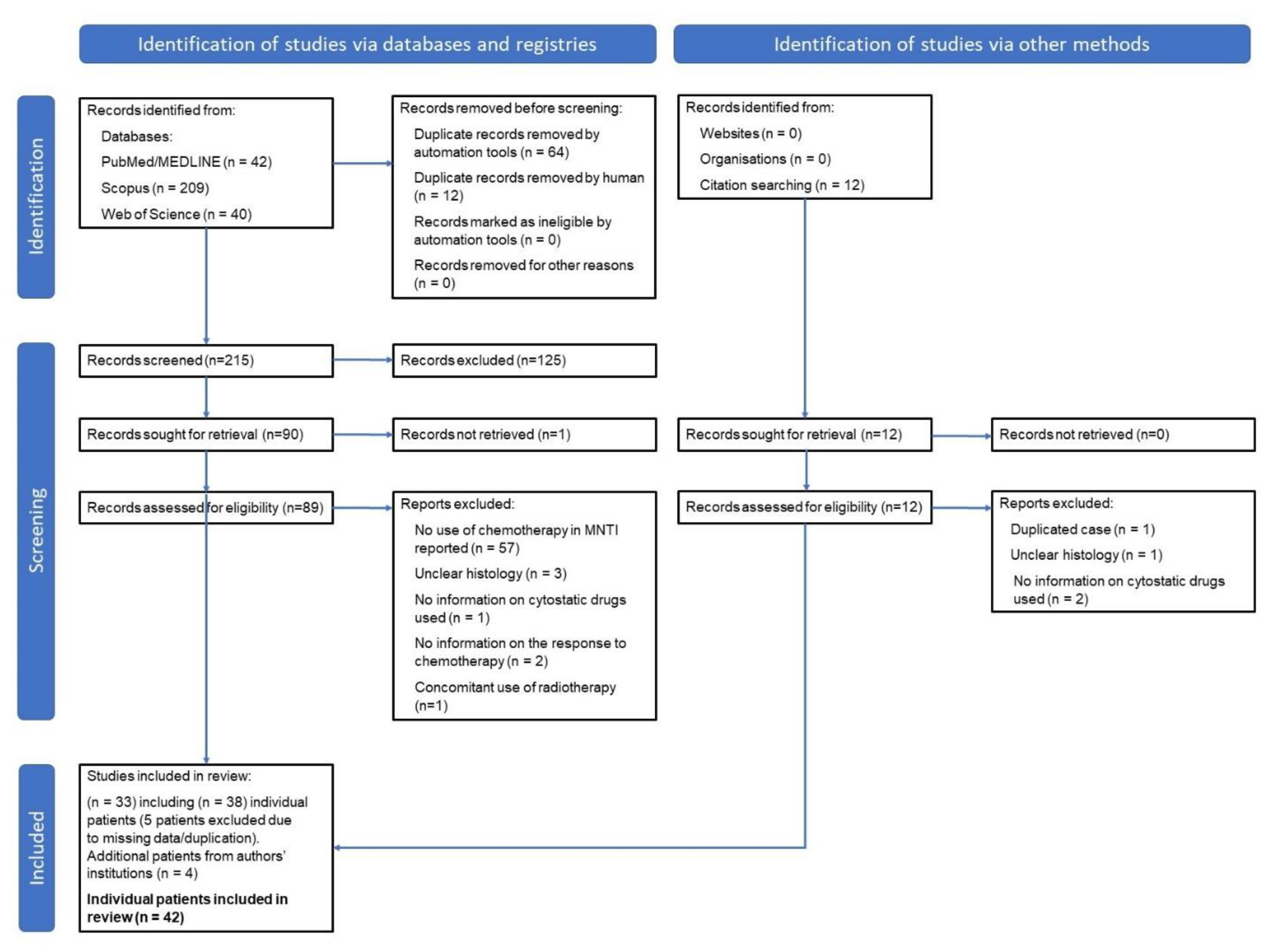
3.1. Methodology
3.2. Results
3.2.1. Chemotherapy in the First-Line Treatment of MNTI
Adjuvant CHT in the First-Line Treatment of MNTI
Neoadjuvant CHT in the First-Line Treatment of MNTI
CHT as the Only First-Line Treatment of MNTI
3.2.2. Chemotherapy in the Treatment of Recurrence of MNTI
Adjuvant CHT in the Treatment of Recurrence of MNTI
Neoadjuvant CHT in the Treatment of Recurrence of MNTI
4. Discussion
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rachidi, S.; Sood, A.J.; Patel, K.G.; Nguyen, S.A.; Hamilton, H.; Neville, B.W.; Day, T.A. Melanotic neuroectodermal tumor of infancy: A systematic review. J. Oral Maxillofac. Surg. 2015, 73, 1946–1956. [Google Scholar] [CrossRef]
- Chaudhary, A.; Wakhlu, A.; Mittal, N.; Misra, S.; Mehrotra, D.; Wakhlu, A.K. Melanotic Neuroectodermal Tumor of Infancy: 2 Decades of Clinical Experience With 18 Patients. J. Oral Maxillofac. Surg. 2009, 67, 47–51. [Google Scholar] [CrossRef]
- Rekhi, B.; Suryavanshi, P.; Desai, S.; Gulia, A.; Desai, S.; Juvekar, S.L.; Puri, A.; Jambhekar, N.A. Melanotic neuroectodermal tumor of infancy in thigh of an infant—A rare case report with diagnostic implications. Skelet. Radiol. 2011, 40, 1079–1084. [Google Scholar] [CrossRef]
- Creytens, D.; Ferdinande, L.; Lecoutere, E.; Van Dorpe, J. Melanotic neuroectodermal tumour of infancy presenting as an undifferentiated round cell tumour in the soft tissue of the forearm. Pathology 2017, 49, 87–90. [Google Scholar] [CrossRef]
- Ghersin, Z.J.; Kuo, D.J. Melanotic Neuroectodermal Tumor of Infancy in the Epididymis: A Brief Report and Review of the Role of Chemotherapy in Management. J. Pediatr. Hematol. Oncol. 2016, 38, e144–e146. [Google Scholar] [CrossRef] [PubMed]
- Burton, K.R.; Ngan, B.Y.; Navarro, O.M. Epididymal melanotic neuroectodermal tumor of infancy: A rare cause of scrotal mass in an infant. J. Clin. Ultrasound 2019, 47, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Ellsworth, B.D.; Young, L.W.; Kheradpour, A.; Zuppan, C.W. Spontaneous Regression of Diffuse Periosteal Melanotic Neuroectodermal Tumor of Infancy in the Tibia, With 13-year Follow-up. J. Pediatr. Hematol. Oncol. 2019, 41, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Kirigin, M.S.; Džombeta, T.; Seiwerth, S.; Mesić, M.; Stepan Giljević, J.; Krušlin, B. Melanotic Neuroectodermal Tumor of Infancy of the Upper Arm. Med. Princ. Pract. 2017, 26, 582–585. [Google Scholar] [CrossRef]
- Choy, J.; Abouzari, M.; Mahboubi, H.; Linskey, M.E.; Djalilian, H.R. Melanotic Neuroectodermal Tumor Presenting as Endolymphatic Sac Tumor. Ear Nose Throat J. 2019, 98, 537–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Li, M.; Tang, X.; Xiao, Y.; Xiao, Z.; Li, Y. Melanotic neuroectodermal tumor of infancy in ovary: A rare case report. Medicine 2019, 98. [Google Scholar] [CrossRef] [PubMed]
- Lurie, H.I. Congenital melanocarcinoma, melanotic adamantinoma, retinal anlage tumor, progonoma, and pigmented epulis of infancy. Cancer 1961, 14, 1090–1108. [Google Scholar] [CrossRef]
- Nikai, H.; Ijuhin, N.; Yamasaki, A.; Nutani, K.; Imai, K. Ultrastructural Evidence for Neural Crest Origin of the Melanotic Neuroectodermal Tumor of Infancy. J. Oral Pathol. Med. 1977, 6, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, S.B.; Frisman, D.M.; Hitchcock, C.L.; Popek, E.J. Melanotic Neuroectodermal Tumor of Infancy. Clinicopathological, Immunohistochemical, and Flow Cytometric Study. Am. J. Surg. Pathol. 1993, 17, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Soles, B.S.; Wilson, A.; Lucas, D.R.; Heider, A. Melanotic neuroectodermal tumor of infancy. Arch. Pathol. Lab. Med. 2018, 142, 1358–1363. [Google Scholar] [CrossRef]
- Barrett, A.W.; Morgan, M.; Ramsay, A.D.; Farthing, P.M.; Newman, L.; Speight, P.M. A clinicopathologic and immunohistochemical analysis of melanotic neuroectodermal tumor of infancy. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2002, 93, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Galmiche, L.; Minard-Colin, V.; Rachwalski, M.; Belhous, K.; Orbach, D.; Joly, A.; Picard, A.; Kadlub, N. Melanotic neuroectodermal tumor of infancy (MNTI) of the head and neck: A French multicenter study. J. Cranio-Maxillo-Facial Surg. 2018, 46, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Enriquez, A.M.; Carnate, J.M., Jr. Melanotic neuroectodermal tumor of infancy. Philipp. J. Otolaryngol. Neck Surg. 2011, 26, 51–54. [Google Scholar] [CrossRef]
- Chrcanovic, B.R.; Gomez, R.S. Melanotic neuroectodermal tumour of infancy of the jaws: An analysis of diagnostic features and treatment. Int. J. Oral Maxillofac. Surg. 2019, 48, 1–8. [Google Scholar] [CrossRef] [Green Version]
- El-Naggar, A.; Chan, J.; Grandis, J.; Takata, T.; Slootweg, P. WHO Classification of Head and Neck Tumors, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; ISBN 978-92-832-2438-9. [Google Scholar]
- Jenkinson, H.; Grundy, R. Guidelines for the Management of Melanotic Neuroectodermal Tumour of Infancy; Children’s Cancer and Leukaemia Group, Study Group Rare Tumour Committee: Leicester, United Kingdom, 2004. [Google Scholar]
- Yoo, I.H.; Yum, S.K.; Oh, S.-J.J.; Kim, K.-M.M.; Jeong, D.C. Melanotic neuroectodermal tumor of infancy disseminated by a ventriculoperitoneal shunt and diagnosed from the inguinal sac. J. Pediatr. Hematol. Oncol. 2014, 36, 61–64. [Google Scholar] [CrossRef] [Green Version]
- Azari, A.; Petrisor, D.; Wright, J.; Ghali, G.E. Metastatic Melanotic Neuroectodermal Tumor of Infancy: Report of a Case and Review of the Literature. J. Oral Maxillofac. Surg. 2016, 74, 2431–2440. [Google Scholar] [CrossRef]
- Furtado, S.V.; Ghosal, N.; Hegde, A.S. Calvarial malignant melanotic neuroectodermal tumour of infancy presenting with widespread intracranial metastasis. J. Cranio-Maxillo-Facial Surg. 2012, 40, e170–e173. [Google Scholar] [CrossRef]
- Pereira, A.A.C.; de Jesus Rozante, M.M.; Bauer Doveinis, R.; Porcelli Salvarani, C.; Hissa Anegawa, T.; da Costa Souza, P.; Brat, D.J.; de Oliveira Borges, A.C. The recurrence of the melanotic neuroectodermal tumour of infancy: An unusual presentation of a rare tumour. Ecancermedicalscience 2020, 14. [Google Scholar] [CrossRef]
- Kruse-Lösler, B.; Gaertner, C.; Bürger, H.; Seper, L.; Joos, U.; Kleinheinz, J. Melanotic neuroectodermal tumor of infancy: Systematic review of the literature and presentation of a case. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2006, 102, 204–216. [Google Scholar] [CrossRef]
- Mosby, E.L.; Lowe, M.W.; Cobb, C.M.; Ennis, R.L. Melanotic Neuroectodermal Tumor of Infancy: Review of the Literature and Report of a Case. J. Oral Maxillofac. Surg. 1992, 50, 886–894. [Google Scholar] [CrossRef]
- Cutler, L.S.; Chaudhry, A.P.; Topazian, R. Melanotic Neuroectodermal Tumor of Infancy: An Ultrastructural Study, Literature Review, and Reevaluation. Cancer 1981, 48, 257–270. [Google Scholar] [CrossRef]
- Block, J.C.; Waite, D.E.; Dehner, L.P.; Leonard, A.S.; Ogle, R.G.; Gatto, D.J. Pigmented neuroectodermal tumor of infancy. An example of rarely expressed malignant behavior. Oral Surg. Oral Med. Oral Pathol. 1980, 49, 279–285. [Google Scholar] [CrossRef]
- Mirich, D.R.; Blaser, S.I.; Harwood-Nash, D.C.; Armstrong, D.C.; Becker, L.E.; Posnick, J.C. Melanotic Neuroectodermal Tumor of Infancy: Clinical, Radiologic, and Pathologic Findings in Five Cases. Am. J. Neuroradiol. 1991, 12, 689–697. [Google Scholar] [PubMed]
- Nadarajah, J.; Garg, A.; Bohara, S.; Garg, K.; Devaranjan Sebastian, L.J.; Suri, V.; Bakhshi, S.; Singh, M. Calvarial Melanotic Neuroectodermal Tumor of Infancy with Rhabdomyosarcomatous differentiation—A Rare Case. World Neurosurg. 2021, 145, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, J.; Chuckwulobelu, R.; Sebire, N.J.; Hartley, B.E.J.; Dunaway, D.J. Hemimandibulectomy and autologous costochondral rib graft reconstruction for a case of melanotic neuroectodermal tumour of infancy arising within the mandible. Int. J. Pediatr. Otorhinolaryngol. Extra 2007, 2, 189–193. [Google Scholar] [CrossRef]
- Sobel, N.; Carcangiu, M.L. Primary Pigmented Neuroectodermal Tumor of the Uterine Cervix. Int. J. Surg. Pathol. 1994, 2, 31–36. [Google Scholar] [CrossRef]
- Ogata, A.; Fujioka, Y.; Nagashima, K.; Tashiro, K.; Aida, T.; Abe, H. Malignant melanotic neuroectodermal tumor arising from the pineal body. Acta Neuropathol. 1989, 77, 654–658. [Google Scholar] [CrossRef]
- Johnson, R.E.; Scheithauer, B.W.; Dahlin, D.C. Melanotic Neuroectodermal Tumor of Infancy. A Review of Seven Cases. Cancer 1983, 52, 661–666. [Google Scholar] [CrossRef]
- Naidoo, J.; Potgieter, L.; Wieselthaler, N.; Pillay, K. Melanotic neuroectodermal tumour of infancy, a rare cause for a bump on the head. Child’s Nerv. Syst. 2013, 29, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Rickart, A.J.; Drummond-Hay, V.; Suchak, A.; Sadiq, Z.; Sebire, N.J.; Slater, O.; Mills, C. Melanotic neuroectodermal tumour of infancy: Refining the surgical approach. Int. J. Oral Maxillofac. Surg. 2019, 48, 1307–1312. [Google Scholar] [CrossRef]
- Hojan-Jezierska, D.; Chomiak, A.; Czopor, A.; Matthews-Kozanecka, M.; Majewska, A.; Urbaniak-Olejnik, M.; Matthews-Brzozowska, T. Ototoxicity after platinum-based chemotherapy in the treatment of melanotic neuroectodermal tumour of infancy. Oncol. Lett. 2020, 19, 3411–3416. [Google Scholar] [CrossRef]
- Ebel, F.; Thieringer, F.M.; Kunz, C.; Klein-Franke, A.; Scheinemann, K.; Guzman, R.; Soleman, J. Melanotic neuroectodermal tumor of infancy to the skull: Case-based review. Child’s Nerv. Syst. 2020, 36, 679–688. [Google Scholar] [CrossRef]
- Haque, S.; McCarville, M.B.; Sebire, N.; McHugh, K. Melanotic neuroectodermal tumour of infancy: CT and MR findings. Pediatr. Radiol. 2012, 42, 699–705. [Google Scholar] [CrossRef] [Green Version]
- Cohen, B.H.; Handler, M.S.; DeVivo, D.C.; Garvin, J.H.; Hays, A.P.; Carmel, P. Central nervous system melanotic neuroectodermal tumor of infancy: Value of chemotherapy in management. Neurology 1988, 38, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, S.; Takahashi, H.; Shimura, T.; Nakazawa, S.; Naito, Z.; Asano, G. Melanotic neuroectodermal tumor of infancy in the skull associated with high serum levels of catecholamine. J. Neurosurg. 1994, 80, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Patankar, T.; Prasad, S.; Goel, A.; Perumpillichira, J.; Desai, A.P. Malignant melanotic neuroectodermal tumour of infancy affecting the occipital squama. J. Postgrad. Med. 1998, 44, 73–75. [Google Scholar]
- Kumari, T.P.; Venugopal, M.; Mathews, A.; Kusumakumary, P. Effectiveness of chemotherapy in melanotic neurectodermal tumor of infancy. Pediatr. Hematol. Oncol. 2005, 22, 199–206. [Google Scholar] [CrossRef]
- Murphy, C.; Pears, J.; Kearns, G.J. Melanotic neuroectodermal tumour of infancy: Surgical and chemotherapeutic management. Ir. J. Med. Sci. 2016, 185, 753–756. [Google Scholar] [CrossRef]
- Higashi, K.; Ogawa, T.; Onuma, M.; Usubuchi, H.; Imai, Y.; Takata, I.; Hidaka, H.; Watanabe, M.; Sasahara, Y.; Koyama, S.; et al. Clinicopathological features of melanotic neuroectodermal tumor of infancy: Report of two cases. Auris Nasus Larynx 2016, 43, 451–454. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, A.; Van Tornout, J.M.; Hachitanda, Y.; Ortega, J.A.; Shimada, H. Melanotic Neuroectodermal Tumor of Infancy. A Case Report of Paratesticular Primary with Lymph Node Involvment. Am. J. Pediatr. Hematol. Oncol. 1992, 14, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Mello, R.J.V.; Vidal, A.K.L.; Fittipaldi, H.M.; Montenegro, L.T.; Calheiros, L.M.C.; Rocha, G.I. Melanotic Neuroectodermal Tumor of Infancy: Clinicopathologic Study of a Case, with Emphasis on the Chemotherapeutic Effects. Int. J. Surg. Pathol. 2000, 8, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Hered, R.W.; Smithwick IV, W.; Sandler, E.; Goldstein, J.D. Orbital melanotic neuroectodermal tumor of infancy successfully treated with chemotherapy and subtotal excision. J. AAPOS 2007, 11, 504–505. [Google Scholar] [CrossRef]
- Choi, I.S.; Kook, H.; Han, D.K.; Baek, H.J.; Jung, S.T.; Lee, J.H.; Park, J.G.; Hwang, T.J. Melanotic Neuroectodermal Tumor of Infancy in the Femur: A Case Report and Review of the Literature. J. Pediatr. Hematol. Oncol. 2007, 29, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Maroun, C.; Khalifeh, I.; Alam, E.; Akl, P.A.; Saab, R.; Moukarbel, R.V. Mandibular melanotic neuroectodermal tumor of infancy: A role for neoadjuvant chemotherapy. Eur. Arch. Oto-Rhino-Laryngol. 2016, 273, 4629–4635. [Google Scholar] [CrossRef]
- Nicosia, G.; Spennato, P.; Aliberti, F.; Cascone, D.; Quaglietta, L.; Errico, M.E.; Muto, M.; Ionna, F.; Cinalli, G. Giant melanotic neuroectodermal tumor of infancy (melanotic progonoma) of the head and neck: Report of a malignant case. J. Neurosurg. Pediatr. 2017, 19, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Khemiri, S.; Feki, J.; Khanfir, A.; Abdelmoula, M.; Frikha, M. Melanotic Neuroectodermal Tumor of Infancy: Presentation of a Case Affecting the Mandible. Acta Med. Iran. 2019, 57, 143–146. [Google Scholar] [CrossRef]
- Pierre-Kahn, A.; Cinalli, G.; Lellouch-Tubiana, A.; Villarejo, F.J.; Sainte-Rose, C.; Pfister, A.; Couly, G. Melanotic Neuroectodermal Tumor of the Skull and Meninges in Infancy. Pediatr. Neurosurg. 1992, 18, 6–15. [Google Scholar] [CrossRef]
- Moreau, A.; Galmiche, L.; Belhous, K.; Franchi, G.; Couloigner, V.; Nevoux, J.; Aymard, P.A.; Picard, A.; Minard-Colin, V.; Kadlub, N. Prenatal Diagnosis of a Melanotic Neuroectodermal Tumor of Infancy (MNTI): A Case Report with a Favorable Outcome after Chemotherapy Failure and Incomplete Resection. J. Pediatr. Hematol. Oncol. 2018, 40, 320–324. [Google Scholar] [CrossRef]
- Woessmann, W.; Neugebauer, M.; Gossen, R.; Blütters-Sawatzki, R.; Reiter, A. Successful chemotherapy for melanotic neuroectodermal tumor of infancy in a baby. Med. Pediatr. Oncol. 2003, 40, 198–199. [Google Scholar] [CrossRef]
- Sailukar, M.; Bhagwat, R.; Seth, T. Melanocytic neuroectodermal tumor of infancy. J. Indian Assoc. Pediatr. Surg. 2007, 12, 136–137. [Google Scholar] [CrossRef]
- Blank, E.; Runckel, D.N. Case Report 119. Skeletal Radiol. 1980, 5, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Shokry, A.; Briner, J.; Makek, M. Malignant Melanotic Neuroectodermal Tumor of Infancy: A Case Report. Pediatr. Pathol. 1986, 5, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Atkinson Jr, G.O.; Davis, P.C.; Patrick, L.E.; Winn, K.J.; Ball, T.I.; Wyly, J.B. Melanotic neuroectodermal tumor of infancy. MR findings and a review of the literature. Pediatr. Radiol. 1989, 20, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Shaia, W.T.; DiNardo, L.J.; Underhill, T.E.; Cesca, C.E. Recurrent melanotic neuroectodermal tumor of infancy. Am. J. Otolaryngol. 2002, 23, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Neven, J.; Hulsbergen-van der Kaa, C.; Groot-Loonen, J.; de Wilde, P.C.M.; Merkx, M.A.W. Recurrent melanotic neuroectodermal tumor of infancy: A proposal for treatment protocol with surgery and adjuvant chemotherapy. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2008, 106, 493–496. [Google Scholar] [CrossRef]
- Davis, J.M.; DeBenedictis, M.; Frank, D.K.; Lessin, M.E. Melanotic Neuroectodermal Tumor of Infancy: A Wolf in Sheep’s Clothing. Ann. Otol. Rhinol. Laryngol. 2015, 124, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Dehner, L.P.; Sibley, R.K.; Sauk, J.J.; Vickers, R.A.; Nesbit, M.E.; Leonard, A.S.; Waite, D.E.; Neeley, J.E.; Ophoven, J. Malignant Melanotic Neuroectodermal Tumor of Infancy. A Clinical, Pathologic, Ultrastructural and Tissue Culture Study. Cancer 1979, 43, 1389–1410. [Google Scholar] [CrossRef]
- Riggi, N.; Suva, M.L.; Stamenkovic, I. Ewing’s sarcoma origin: From duel to duality. Expert Rev. Anticancer Ther. 2009, 9, 1025–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marschall, J.S.; Kushner, G.M.; Shumway, B.S. Agressive histologic features do not predict biologic behavior in melanotic neuroectodermal tumor of infancy. J. Oral Maxillofac. Pathol. 2018, 22, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.J.; Hookway, E.; Athanasou, N.; Kashima, T.; Oppermann, U.; Hughes, S.; Swan, D.; Lueerssen, D.; Anson, J.; Hassan, A.B. A germline mutation of CDKN2A and a novel RPLP1-C19MC fusion detected in a rare melanotic neuroectodermal tumor of infancy: A case report. BMC Cancer 2016, 16, 629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, C.C.; Diniz, M.G.; de Menezes, G.H.F.; Castro, W.H.; Gomez, R.S. BRAFV600E Mutation in Melanotic Neuroectodermal Tumor of Infancy: Toward Personalized Medicine? Pediatrics 2015, 136, e267–e269. [Google Scholar] [CrossRef] [Green Version]
- Hoshina, Y.; Hamamoto, Y.; Suzuki, I.; Nakajima, T.; Ida-Yonemochi, H.; Saku, T. Melanotic neuroectodermal tumor of infancy in the mandible: Report of a case. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2000, 89, 594–599. [Google Scholar] [CrossRef]
- Judd, P.L.; Pedod, D.; Harrop, K.; Becker, J. Melanotic neuroectodermal tumor of infancy. Oral Surg. Oral Med. Oral Pathol. 1990, 69, 723–726. [Google Scholar] [CrossRef]
- Pettinato, G.; Manivel, J.C.; D’Amore, E.S.G.; Jaszcz, W.; Gorlin, R.J. Melanotic neuroectodermal tumor of infancy: A reexamination of a histogenetic problem based on immunohistochemical, flow cytometric, and ultrastructural study of 10 cases. Am. J. Surg. Pathol. 1991, 15, 233–245. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Treatment | Cytostatic Drugs | Number of Courses | Response |
|---|---|---|---|
| neoadjuvant | CADO (cyclophosphamide, vincristine, doxorubicin) | 1 | stable disease (tumor growth stopped) |
| neoadjuvant | VP-16 + Carbo (etoposide + carboplatin) | 2 | minor partial response → slow tumor progression |
| neoadjuvant | I2VAdr (ifosfamide, vincristine, doxorubicin with dose reduction to 3/4) | 1 | stable disease |
| neoadjuvant | I2VE (ifosfamide with dose reduction to 2/3, vincristine, etoposide) | 1 | stable disease |
| neoadjuvant | CADO (cyclophosphamide, vincristine, doxorubicin) | 1 | stable disease |
| neoadjuvant | VP-16 + Carbo (etoposide + carboplatin) | 1 | minor partial response |
| neoadjuvant | CO (vincristine, cyclophosphamide) | 1 | minor partial response |
| Type of the Treatment | Cytostatic Drugs | Number of Courses | Response |
|---|---|---|---|
| neoadjuvant | VIDE (vincristine, ifosfamide, doxorubicin, etoposide) | 6 | partial response |
| microscopically incomplete (R1) resection of the tumor | |||
| adjuvant | VAC (vincristine, dactinomycin cyclophosphamide) | 8 | no recurrence |
| ALL treatment (chemotherapy + allo-HSCT) | vincristine, daunorubicin, L-asparaginase, cyclophosphamide, ifosfamid, cytarabine, inthrathecal cytarabine, intrathecal metothrexate, doxorubicin, 6-mercaptopurin, 6-tioguanine, vindesine, etopozide | >2 years of treatment | no recurrence of ALL and MNTI |
| Type of the Treatment | Cytostatic Drugs | Number of Courses | Response |
|---|---|---|---|
| neoadjuvant | N4 (vincristine, cyclophosphamide, doxorubicin) | 1 | stable disease |
| neoadjuvant | N4 (vincristine, cyclophosphamide, doxorubicin—accidental overdosage) | 1 | stable disease→watch&wait→slow progression |
| partial resection of the tumor | |||
| adjuvant | N5 (cisplatin, etoposide, vindesine) | 1 | not assessed |
| adjuvant | N6 (vincristine, dacarbazin, ifosfamide, without doxorubicin) | 1 | stable disease |
| adjuvant | N5 (cisplatin, etoposide, vindesine) | 1 | stable disease→follow-up →progession |
| Type of the Treatment | Cytostatic Drugs | Number of Courses | Response |
|---|---|---|---|
| neoadjuvant | N4 (vincristine, cyclophosphamide, doxorubicin) | 4 | stable disease |
| All Patients | n = 42 (100%) |
|---|---|
| own cases | n = 4 (9.5%) |
| literature reports | n = 38 (90.5%) |
| Sex | |
| female | n = 12 (28.6%) |
| male | n = 27 (64.3%) |
| unknown | n = 3 (7.1%) |
| Median age at diagnosis (mo) | 4 (range: 0–48) |
| Tumor site | |
| maxilla | n = 19 (45.2%) |
| skull | n = 9 (21.4%) |
| mandible | n = 5 (11.9%) |
| femur | n = 2 (4.8%) |
| CNS | n = 2 (4.8%) |
| orbit | n = 2 (4.8%) |
| epididymis | n = 1 (2.4%) |
| suboccipital area | n = 1 (2.4%) |
| forearm | n = 1 (2.4%) |
| Median tumor size at diagnosis (cm) | 4 (range: 1–20.5) |
| Metastases at diagnosis | |
| no | n = 12 (28.6%) |
| yes | n = 4 (9.5%) |
| lymph nodes | n = 3 (7.1%) |
| distant metastases | n = 1 (2.4%) |
| unknown | n = 26 (61.9%) |
| Type of CHT | Number of Patients | Response to CHT | Outcome |
|---|---|---|---|
| adjuvant | 10 (41.7%) * | ||
| after macroscopically incomplete (R2) surgery of the primary tumor | 5 | CR: 2 | NED: 2 |
| PR: 1 | NED: 1 | ||
| SD: 2 | AWD: 2* | ||
| after macroscopically incomplete (R2) surgery of the metastatic lesion | 1 | PR: 1 | DOD: 1 |
| after microscopically incomplete (R1) surgery of the primary tumor | 2 | NA: 2 | NED: 2 |
| after complete (R0) surgery of the primary tumor and involved LNs | 2 | NA: 2 | NED: 2 |
| neoadjuvant | 12 (50%) * | ||
| after biopsy of primary tumor | 11 | PR: 7 | NED: 4 D of complications: 1 LFU: 2 |
| SD: 3 | NED: 1 AWD: 1 * LFU: 1 | ||
| PD: 1 | NED: 1 | ||
| after biopsy of involved LN | 1 | PR: 1 | DOD: 1 |
| the only treatment | 3 (12,5%) * | ||
| after biopsy | 3 | PR: 2 | AWD: 2 * |
| SD: 1 | LFU: 1 | ||
| together | 24 (100%) | CR: 2 PR: 12 SD: 4 SD; SD: 1 ** PD: 1 NA: 4 | NED: 13 AWD: 4 * DOD: 2 D of complications: 1 LFU: 4 |
| Type of CHT | Number of Patients | Response to CHT | Outcome |
|---|---|---|---|
| adjuvant | 12 (66.7%) * | ||
| after macroscopically incomplete (R2) surgery of the recurrent tumor | 4 | CR: 1 | NED: 1 * |
| PR: 2 | AWD: 2 | ||
| SD: 1 | AWD: 1 | ||
| after microscopically incomplete (R1) surgery of the recurrent tumor | 5 | NA: 5 | NED: 5* |
| after unspecified surgery of the recurrent tumor | 3 | PD: 2 | DOD: 1 LFU: 1 (in progression) * |
| NA: 1 | NED: 1* | ||
| neoadjuvant | 8 (44.4%) * | ||
| after local recurrence | 6 | CR: 1 | NED: 1* |
| PR: 3 | NED: 3* | ||
| SD: 1 | AWD: 1 | ||
| PD: 1 | NED: 1* | ||
| after regional/metastatic recurrence | 2 | PR: 1 | LFU: 1 (in progression) * |
| PD: 1 | DOD: 1 | ||
| together | 18 (100%) | CR: 2 PR: 2 PR; PD: 1 ** PR; NA: 1 ** SD: 2 PD: 4 NA: 6 | NED: 11 * AWD: 4 DOD: 2 LFU: 1* |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Styczewska, M.; Krawczyk, M.A.; Brecht, I.B.; Haug, K.; Iżycka-Świeszewska, E.; Godziński, J.; Raciborska, A.; Ussowicz, M.; Kukwa, W.; Cwalina, N.; et al. The Role of Chemotherapy in Management of Inoperable, Metastatic and/or Recurrent Melanotic Neuroectodermal Tumor of Infancy—Own Experience and Systematic Review. Cancers 2021, 13, 3872. https://doi.org/10.3390/cancers13153872
Styczewska M, Krawczyk MA, Brecht IB, Haug K, Iżycka-Świeszewska E, Godziński J, Raciborska A, Ussowicz M, Kukwa W, Cwalina N, et al. The Role of Chemotherapy in Management of Inoperable, Metastatic and/or Recurrent Melanotic Neuroectodermal Tumor of Infancy—Own Experience and Systematic Review. Cancers. 2021; 13(15):3872. https://doi.org/10.3390/cancers13153872
Chicago/Turabian StyleStyczewska, Małgorzata, Małgorzata A. Krawczyk, Ines B. Brecht, Konrad Haug, Ewa Iżycka-Świeszewska, Jan Godziński, Anna Raciborska, Marek Ussowicz, Wojciech Kukwa, Natalia Cwalina, and et al. 2021. "The Role of Chemotherapy in Management of Inoperable, Metastatic and/or Recurrent Melanotic Neuroectodermal Tumor of Infancy—Own Experience and Systematic Review" Cancers 13, no. 15: 3872. https://doi.org/10.3390/cancers13153872