
CD14 Expressing Precursors Give Rise to Highly Functional Conventional Dendritic Cells for Use as Dendritic Cell Vaccine

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. CB Collection and CD34 Isolation
2.2. CB-DC Culture
2.3. DC Phenotype
2.4. Uptake Assay
2.5. Mixed Leukocyte Reaction (MLR)
2.6. WT1 Antigen Presentation
2.7. RNA-Sequence
2.8. Statistical Analysis
3. Results
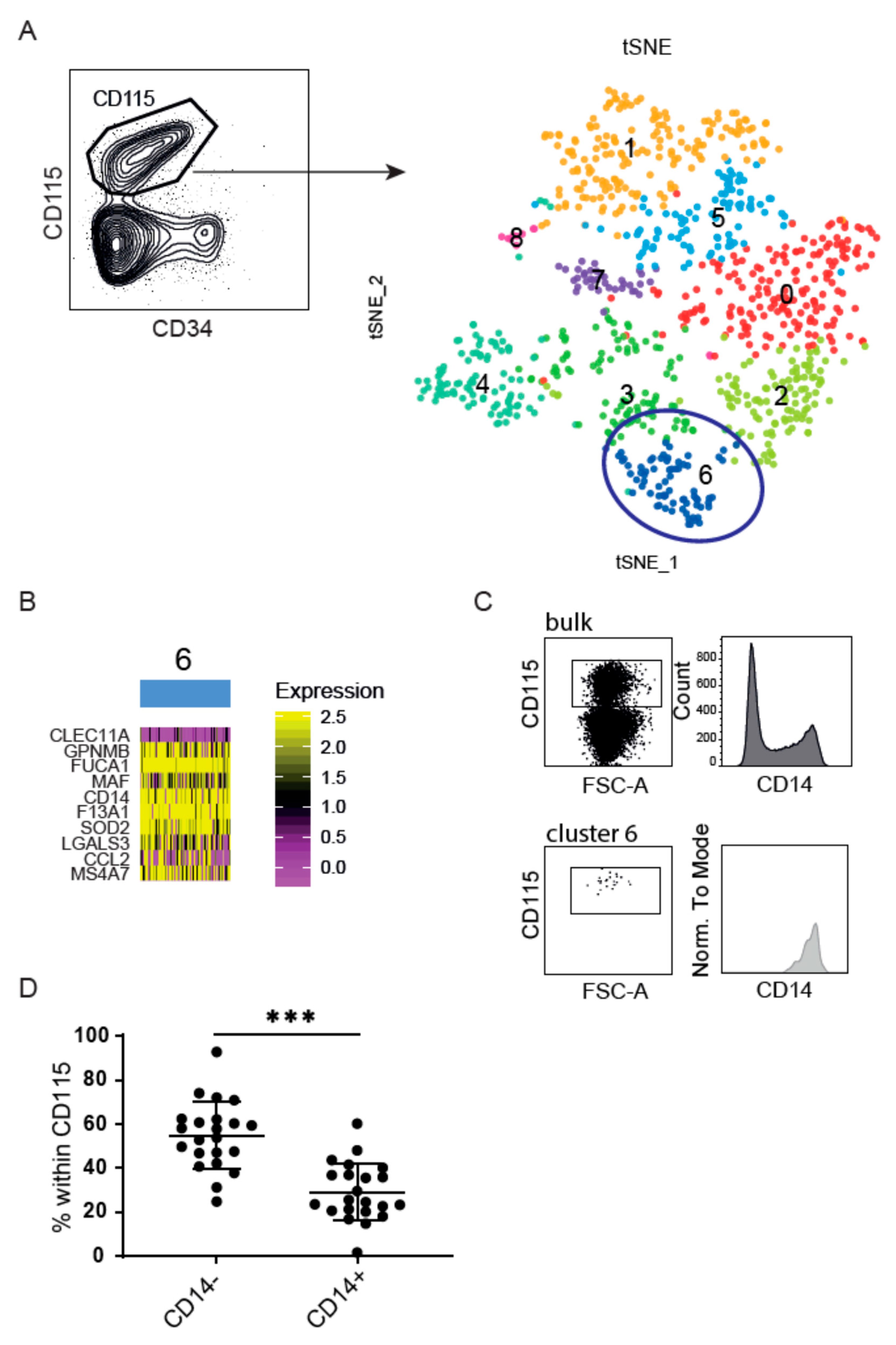
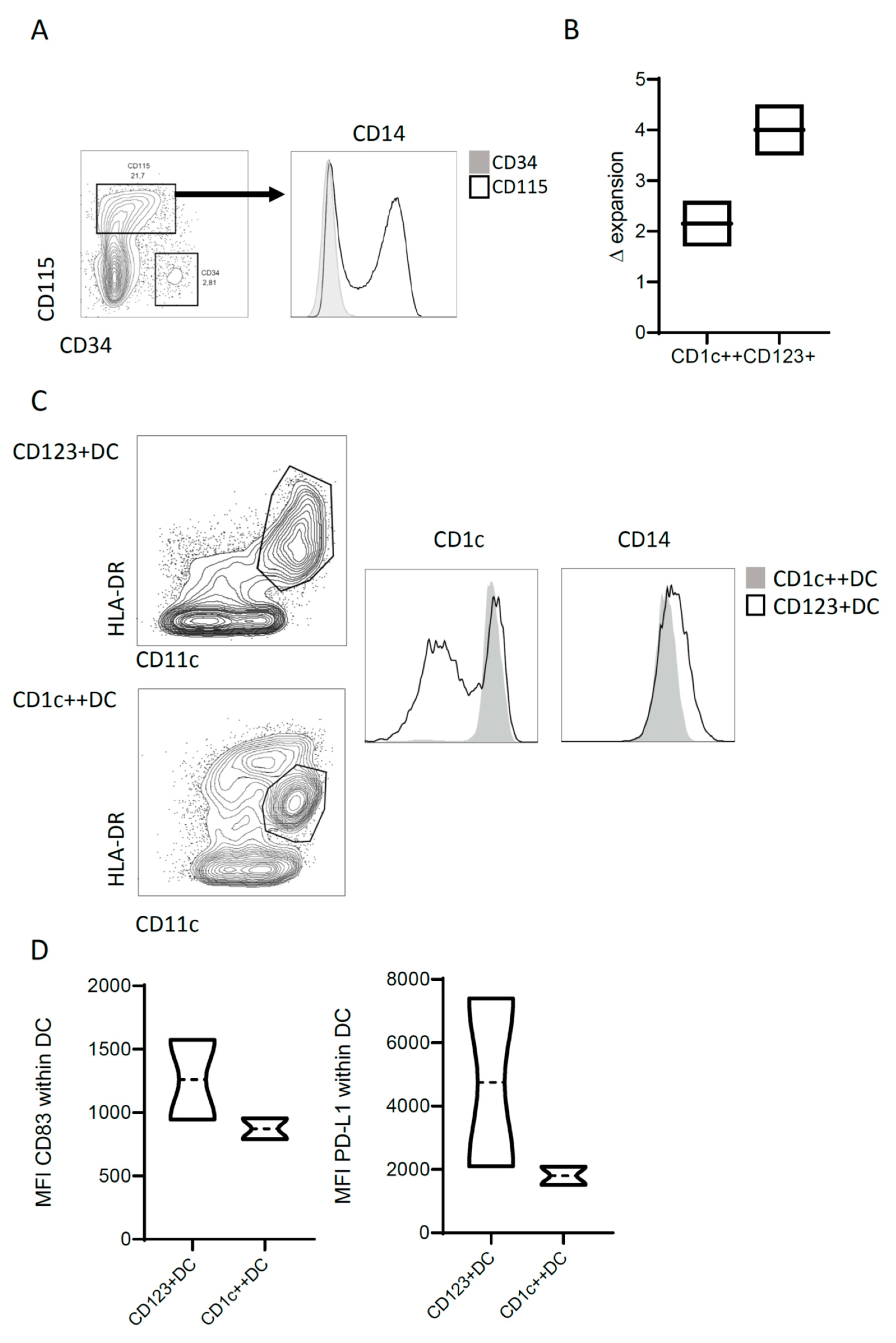
3.1. Heterogenic CD14 Expression within CB-Derived CD115+ DC Precursors
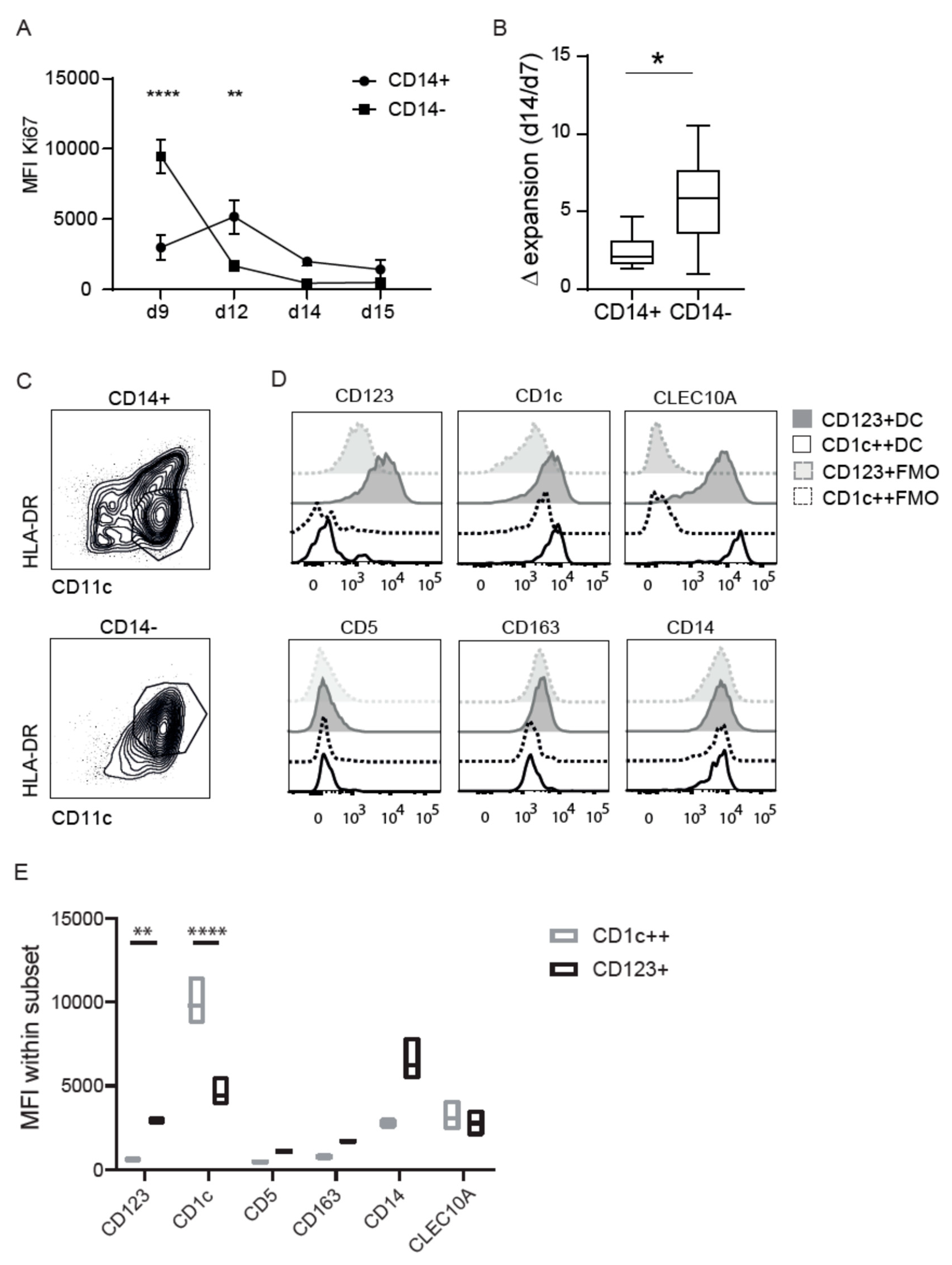
3.2. CD14 Discriminates Two Distinct Differentiated CD1c Populations
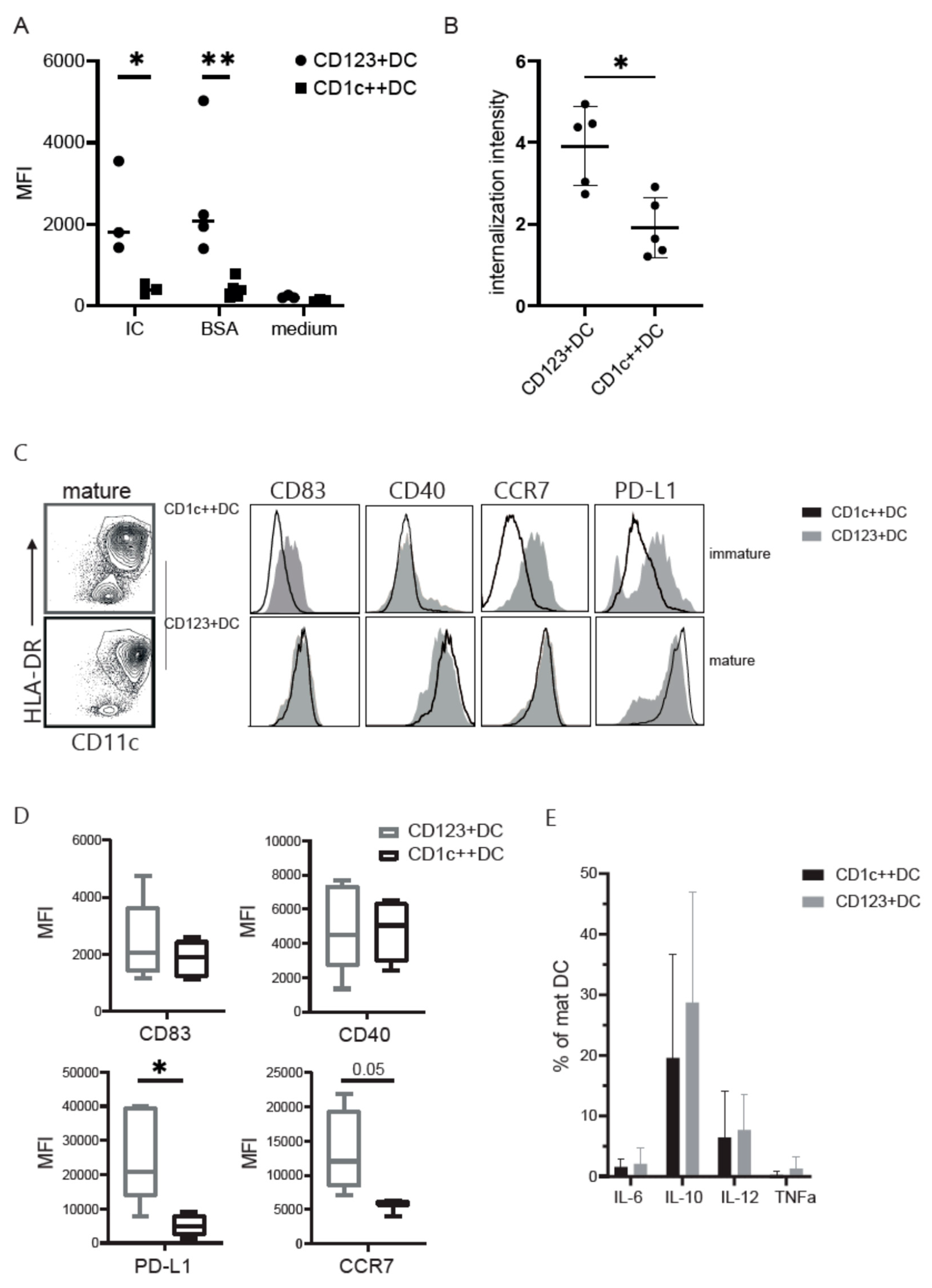
3.3. CD123+DC and CD1c++DC Exhibit Functional Differences
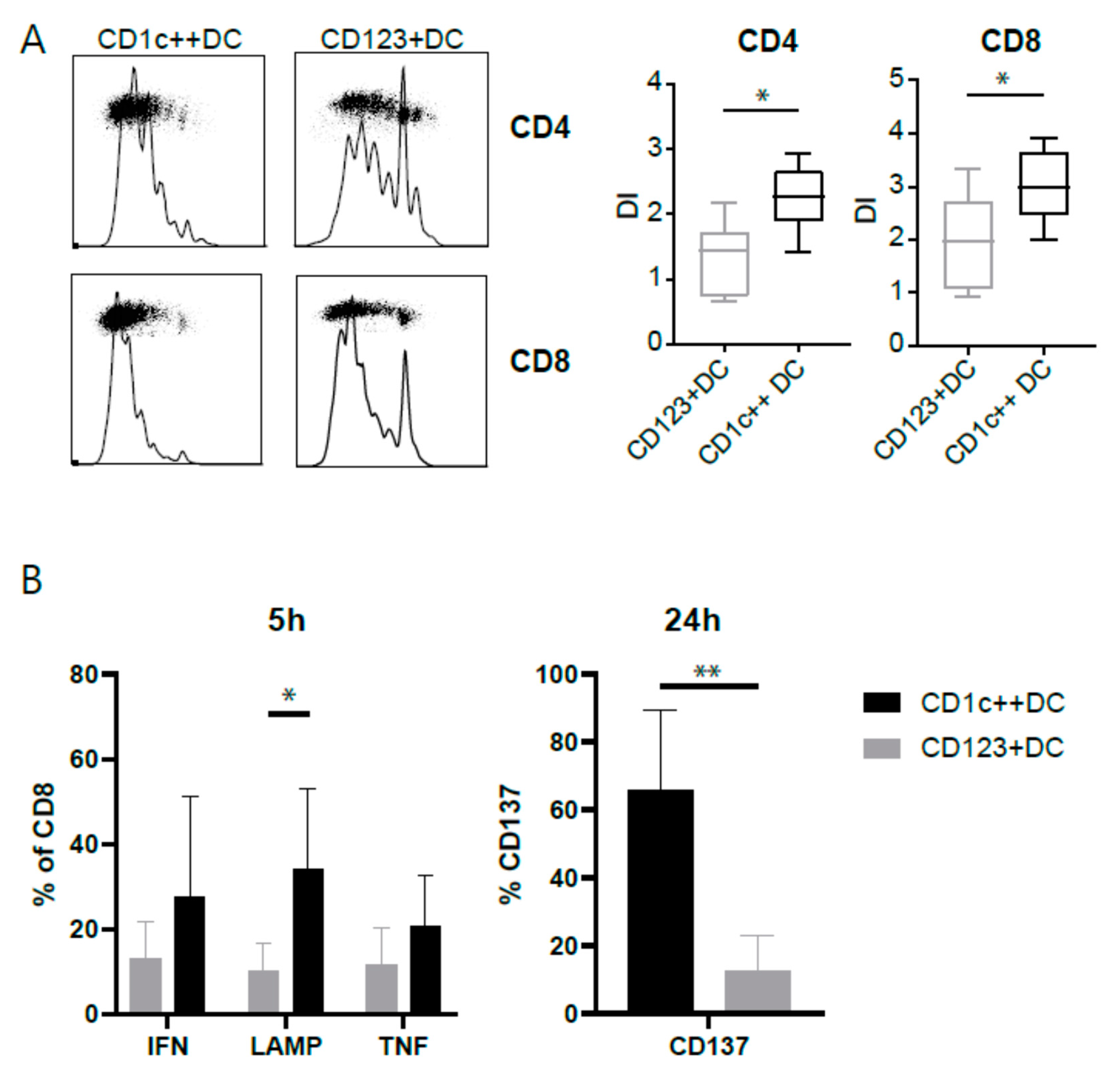
3.4. CD123+DCs and CD1c++DCs Have a Distinct T-Cell Stimulation Capacity
3.5. Translation towards a Clinical Grade Protocol
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; Bhardwaj, N. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer 2018, 4, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Nierkens, S.; Tel, J.; Janssen, E.; Adema, G.J. Antigen cross-presentation by dendritic cell subsets: One general or all sergeants? Trends Immunol. 2013, 34, 361–370. [Google Scholar] [CrossRef] [Green Version]
- van Beek, J.J.P.; Florez-Grau, G.; Gorris, M.A.J.; Mathan, T.S.M.; Schreibelt, G.; Bol, K.F.; Textor, J.; de Vries, I.J.M. Human pDCs Are Superior to cDC2s in Attracting Cytolytic Lymphocytes in Melanoma Patients Receiving DC Vaccination. Cell Rep. 2020, 30, 1027–1038.e4. [Google Scholar] [CrossRef] [Green Version]
- Cytlak, U.; Resteu, A.; Pagan, S.; Green, K.; Milne, P.; Maisuria, S.; McDonald, D.; Hulme, G.; Filby, A.; Carpenter, B.; et al. Differential IRF8 Transcription Factor Requirement Defines Two Pathways of Dendritic Cell Development in Humans. Immunity 2020, 53, 353–370.e8. [Google Scholar] [CrossRef]
- Bourdely, P.; Anselmi, G.; Vaivode, K.; Ramos, R.N.; Missolo-Koussou, Y.; Hidalgo, S.; Tosselo, J.; Nunez, N.; Richer, W.; Vincent-Salomon, A.; et al. Transcriptional and Functional Analysis of CD1c(+) Human Dendritic Cells Identifies a CD163(+) Subset Priming CD8(+)CD103(+) T Cells. Immunity 2020, 53, 335–352.e8. [Google Scholar] [CrossRef]
- Dutertre, C.A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; van den Hoogen, L.L.; Leong, J.Y.; Lee, B.; et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 2019, 51, 573–589.e8. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Poulin, L.F.; Salio, M.; Griessinger, E.; Anjos-Afonso, F.; Craciun, L.; Chen, J.L.; Keller, A.M.; Joffre, O.; Zelenay, S.; Nye, E.; et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010, 207, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Bachem, A.; Guttler, S.; Hartung, E.; Ebstein, F.; Schaefer, M.; Tannert, A.; Salama, A.; Movassaghi, K.; Opitz, C.; Mages, H.W.; et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J. Exp. Med. 2010, 207, 1273–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Yu, H.; Jin, X.; Li, J.; Guo, H.; Shi, Q.; Yin, Z.; Xu, Y.; Wang, X.; Liu, R.; et al. Human Blood CD1c+ Dendritic Cells Encompass CD5high and CD5low Subsets That Differ Significantly in Phenotype, Gene Expression, and Functions. J. Immunol. 2017, 198, 1553–1564. [Google Scholar] [CrossRef] [Green Version]
- Kumamoto, Y.; Linehan, M.; Weinstein, J.S.; Laidlaw, B.J.; Craft, J.E.; Iwasaki, A. CD301b(+) dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity 2013, 39, 733–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tussiwand, R.; Everts, B.; Grajales-Reyes, G.E.; Kretzer, N.M.; Iwata, A.; Bagaitkar, J.; Wu, X.; Wong, R.; Anderson, D.A.; Murphy, T.L.; et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity 2015, 42, 916–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Wilson, N.S.; Waithman, J.; Villadangos, J.A.; Carbone, F.R.; Heath, W.R.; Belz, G.T. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat. Immunol. 2004, 5, 1143–1148. [Google Scholar] [CrossRef]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Heath, W.R.; Carbone, F.R. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat. Immunol. 2009, 10, 1237–1244. [Google Scholar] [CrossRef]
- Pulendran, B.; Banchereau, J.; Burkeholder, S.; Kraus, E.; Guinet, E.; Chalouni, C.; Caron, D.; Maliszewski, C.; Davoust, J.; Fay, J.; et al. Flt3-ligand and granulocyte colony-stimulating factor mobilize distinct human dendritic cell subsets in vivo. J. Immunol. 2000, 165, 566–572. [Google Scholar] [CrossRef] [Green Version]
- Waskow, C.; Liu, K.; Darrasse-Jeze, G.; Guermonprez, P.; Ginhoux, F.; Merad, M.; Shengelia, T.; Yao, K.; Nussenzweig, M. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat. Immunol. 2008, 9, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton, G.; Lee, J.; Zhou, Y.J.; Schreiber, J.J.; Keler, T.; Puhr, S.; Anandasabapathy, N.; Schlesinger, S.; Caskey, M.; Liu, K.; et al. Circulating precursors of human CD1c+ and CD141+ dendritic cells. J. Exp. Med. 2015, 212, 401–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Breton, G.; Oliveira, T.Y.; Zhou, Y.J.; Aljoufi, A.; Puhr, S.; Cameron, M.J.; Sekaly, R.P.; Nussenzweig, M.C.; Liu, K. Restricted dendritic cell and monocyte progenitors in human cord blood and bone marrow. J. Exp. Med. 2015, 212, 382–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balan, S.; Arnold-Schrauf, C.; Abbas, A.; Couespel, N.; Savoret, J.; Imperatore, F.; Villani, A.C.; Vu Manh, T.P.; Bhardwaj, N.; Dalod, M. Large-Scale Human Dendritic Cell Differentiation Revealing Notch-Dependent Lineage Bifurcation and Heterogeneity. Cell Rep. 2018, 24, 1902–1915.e6. [Google Scholar] [CrossRef] [Green Version]
- Bosteels, C.; Scott, C.L. Transcriptional regulation of DC fate specification. Mol. Immunol. 2020, 121, 38–46. [Google Scholar] [CrossRef]
- Plantinga, M.; Lo Presti, V.; de Haar, C.G.; Dunnebach, E.; Madrigal, A.; Lindemans, C.A.; Boelens, J.J.; Nierkens, S. Clinical Grade Production of Wilms’ Tumor-1 Loaded Cord Blood-Derived Dendritic Cells to Prevent Relapse in Pediatric AML After Cord Blood Transplantation. Front. Immunol. 2020, 11, 559152. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, H.; Aronow, B.J.; Jegga, A.G. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinform. 2007, 8, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muraro, M.J.; Dharmadhikari, G.; Grun, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e3. [Google Scholar] [CrossRef] [Green Version]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- de Haar, C.; Plantinga, M.; Blokland, N.J.; van Til, N.P.; Flinsenberg, T.W.; Van Tendeloo, V.F.; Smits, E.L.; Boon, L.; Spel, L.; Boes, M.; et al. Generation of a cord blood-derived Wilms Tumor 1 dendritic cell vaccine for AML patients treated with allogeneic cord blood transplantation. Oncoimmunology 2015, 4, e1023973. [Google Scholar] [CrossRef] [PubMed]
- Plantinga, M.; de Haar, C.G.; Dunnebach, E.; van den Beemt, D.; Bloemenkamp, K.W.M.; Mokry, M.; Boelens, J.J.; Nierkens, S. Cord-Blood-Stem-Cell-Derived Conventional Dendritic Cells Specifically Originate from CD115-Expressing Precursors. Cancers 2019, 11, 181. [Google Scholar] [CrossRef] [Green Version]
- Paul, F.; Arkin, Y.; Giladi, A.; Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Winter, D.; Lara-Astiaso, D.; Gury, M.; Weiner, A.; et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 2015, 163, 1663–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, N.; Fujioka, K.; Denda-Nagai, K.; Hashimoto, S.; Nagai, S.; Sato, T.; Fujita, Y.; Morikawa, A.; Tsuiji, M.; Miyata-Takeuchi, M.; et al. The macrophage C-type lectin specific for galactose/N-acetylgalactosamine is an endocytic receptor expressed on monocyte-derived immature dendritic cells. J. Biol. Chem. 2002, 277, 20686–20693. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.L.; Tai, J.J.; Wong, W.C.; Han, H.; Sem, X.; Yeap, W.H.; Kourilsky, P.; Wong, S.C. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011, 118, e16–e31. [Google Scholar] [CrossRef] [Green Version]
- Heger, L.; Balk, S.; Luhr, J.J.; Heidkamp, G.F.; Lehmann, C.H.K.; Hatscher, L.; Purbojo, A.; Hartmann, A.; Garcia-Martin, F.; Nishimura, S.I.; et al. CLEC10A Is a Specific Marker for Human CD1c(+) Dendritic Cells and Enhances Their Toll-Like Receptor 7/8-Induced Cytokine Secretion. Front. Immunol. 2018, 9, 744. [Google Scholar] [CrossRef]
- Heidkamp, G.F.; Sander, J.; Lehmann, C.H.K.; Heger, L.; Eissing, N.; Baranska, A.; Luhr, J.J.; Hoffmann, A.; Reimer, K.C.; Lux, A.; et al. Human lymphoid organ dendritic cell identity is predominantly dictated by ontogeny, not tissue microenvironment. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef]
- Bakdash, G.; Buschow, S.I.; Gorris, M.A.; Halilovic, A.; Hato, S.V.; Skold, A.E.; Schreibelt, G.; Sittig, S.P.; Torensma, R.; Duiveman-de Boer, T.; et al. Expansion of a BDCA1+CD14+ Myeloid Cell Population in Melanoma Patients May Attenuate the Efficacy of Dendritic Cell Vaccines. Cancer Res. 2016, 76, 4332–4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 2019, 177, 556–571.e16. [Google Scholar] [CrossRef] [PubMed]
- Michea, P.; Noel, F.; Zakine, E.; Czerwinska, U.; Sirven, P.; Abouzid, O.; Goudot, C.; Scholer-Dahirel, A.; Vincent-Salomon, A.; Reyal, F.; et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat. Immunol. 2018, 19, 885–897. [Google Scholar] [CrossRef]
- van Ee, T.J.; Van Acker, H.H.; van Oorschot, T.G.; Van Tendeloo, V.F.; Smits, E.L.; Bakdash, G.; Schreibelt, G.; de Vries, I.J.M. BDCA1+CD14+ Immunosuppressive Cells in Cancer, a Potential Target? Vaccines 2018, 6, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tel, J.; Schreibelt, G.; Sittig, S.P.; Mathan, T.S.; Buschow, S.I.; Cruz, L.J.; Lambeck, A.J.; Figdor, C.G.; de Vries, I.J. Human plasmacytoid dendritic cells efficiently cross-present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood 2013, 121, 459–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, M.C.; Tullett, K.M.; Lee, Y.S.; Idris, A.; Ding, Y.; McDonald, K.J.; Kassianos, A.; Leal Rojas, I.M.; Jeet, V.; Lahoud, M.H.; et al. Differential uptake and cross-presentation of soluble and necrotic cell antigen by human DC subsets. Eur. J. Immunol. 2016, 46, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Carenza, C.; Calcaterra, F.; Oriolo, F.; Di Vito, C.; Ubezio, M.; Della Porta, M.G.; Mavilio, D.; Della Bella, S. Costimulatory Molecules and Immune Checkpoints Are Differentially Expressed on Different Subsets of Dendritic Cells. Front. Immunol. 2019, 10, 1325. [Google Scholar] [CrossRef]
- Corinti, S.; Albanesi, C.; la Sala, A.; Pastore, S.; Girolomoni, G. Regulatory activity of autocrine IL-10 on dendritic cell functions. J. Immunol. 2001, 166, 4312–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Smedt, T.; Van Mechelen, M.; De Becker, G.; Urbain, J.; Leo, O.; Moser, M. Effect of interleukin-10 on dendritic cell maturation and function. Eur. J. Immunol. 1997, 27, 1229–1235. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-10 production by effector T cells: Th1 cells show self control. J. Exp. Med. 2007, 204, 239–243. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plantinga, M.; van den Beemt, D.A.M.H.; Dünnebach, E.; Nierkens, S. CD14 Expressing Precursors Give Rise to Highly Functional Conventional Dendritic Cells for Use as Dendritic Cell Vaccine. Cancers 2021, 13, 3818. https://doi.org/10.3390/cancers13153818
Plantinga M, van den Beemt DAMH, Dünnebach E, Nierkens S. CD14 Expressing Precursors Give Rise to Highly Functional Conventional Dendritic Cells for Use as Dendritic Cell Vaccine. Cancers. 2021; 13(15):3818. https://doi.org/10.3390/cancers13153818
Chicago/Turabian StylePlantinga, Maud, Denise A. M. H. van den Beemt, Ester Dünnebach, and Stefan Nierkens. 2021. "CD14 Expressing Precursors Give Rise to Highly Functional Conventional Dendritic Cells for Use as Dendritic Cell Vaccine" Cancers 13, no. 15: 3818. https://doi.org/10.3390/cancers13153818