
Potent Anticancer Effects of Epidithiodiketopiperazine NT1721 in Cutaneous T Cell Lymphoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Determination of IC50 Values
2.4. Proliferation Assay
2.5. Cell Cycle Analysis
2.6. QPCR
2.7. Western Blots
2.8. In Vivo Studies
2.9. Statistical Analysis
3. Results
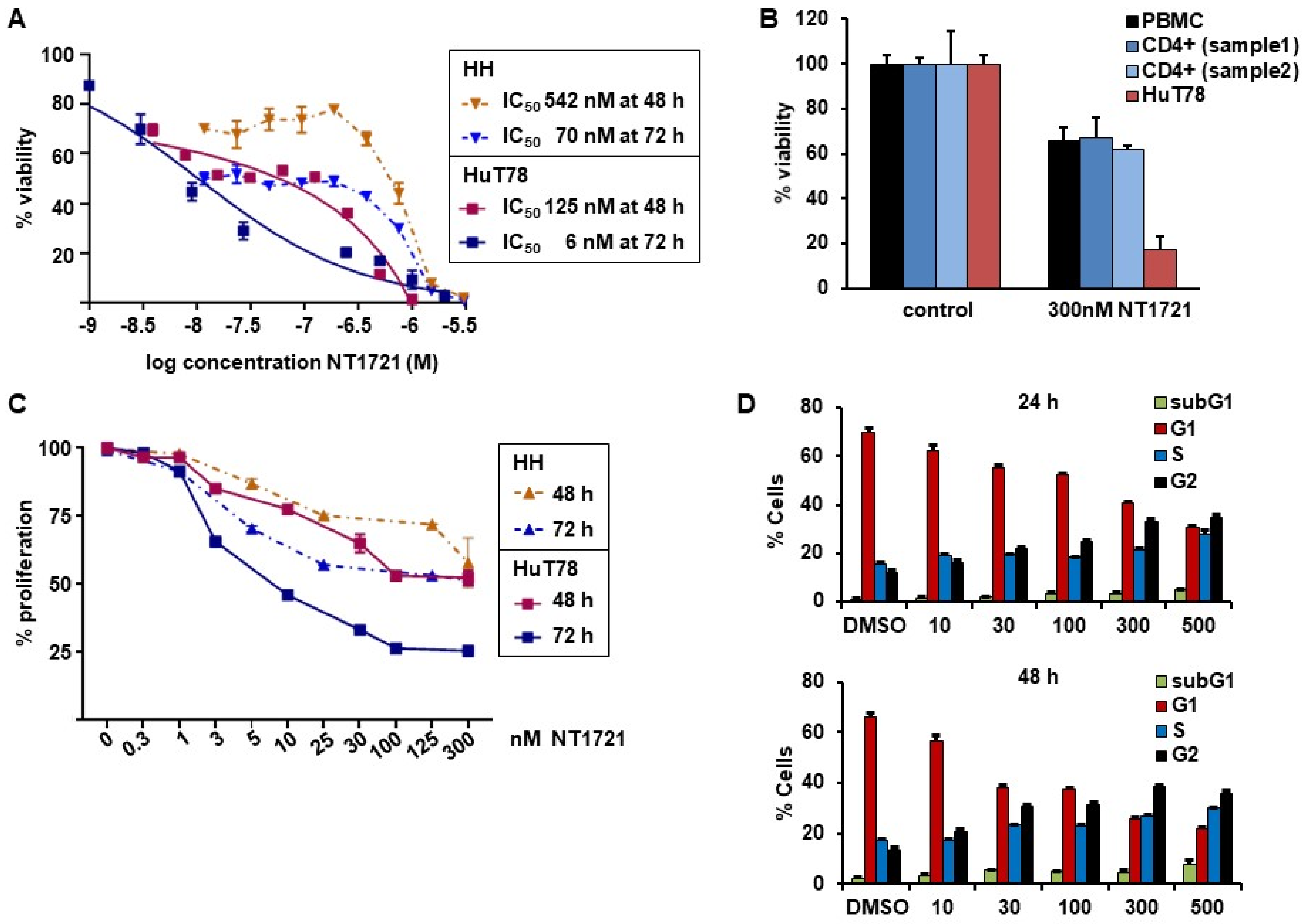
3.1. NT1721 Led to Reduced CTCL Cell Viability, Proliferation and G2 Cell Cycle Arrest at Nanomolar Concentrations
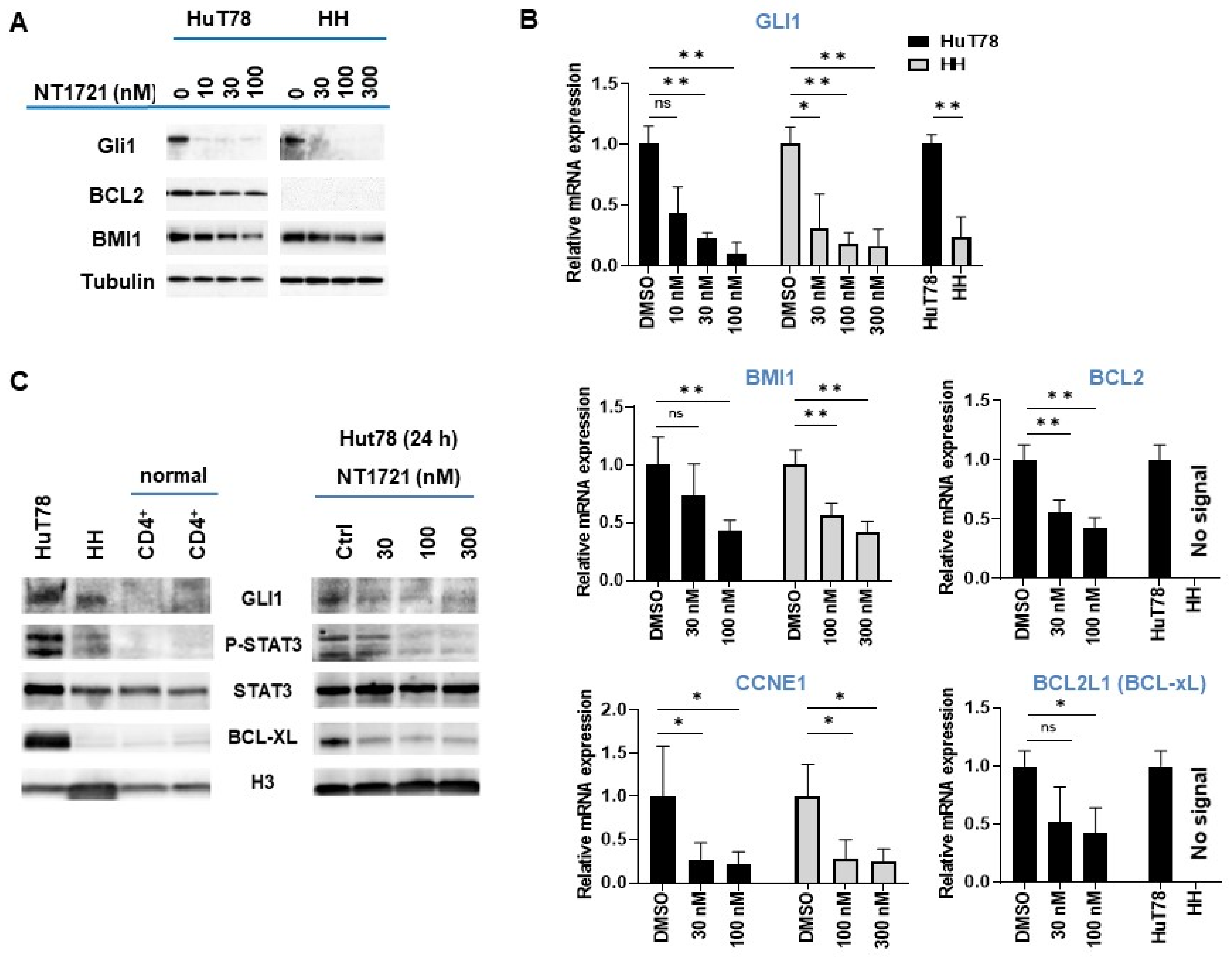
3.2. NT1721 Downregulated GLI1 Transcription Factor in CTCL Cells
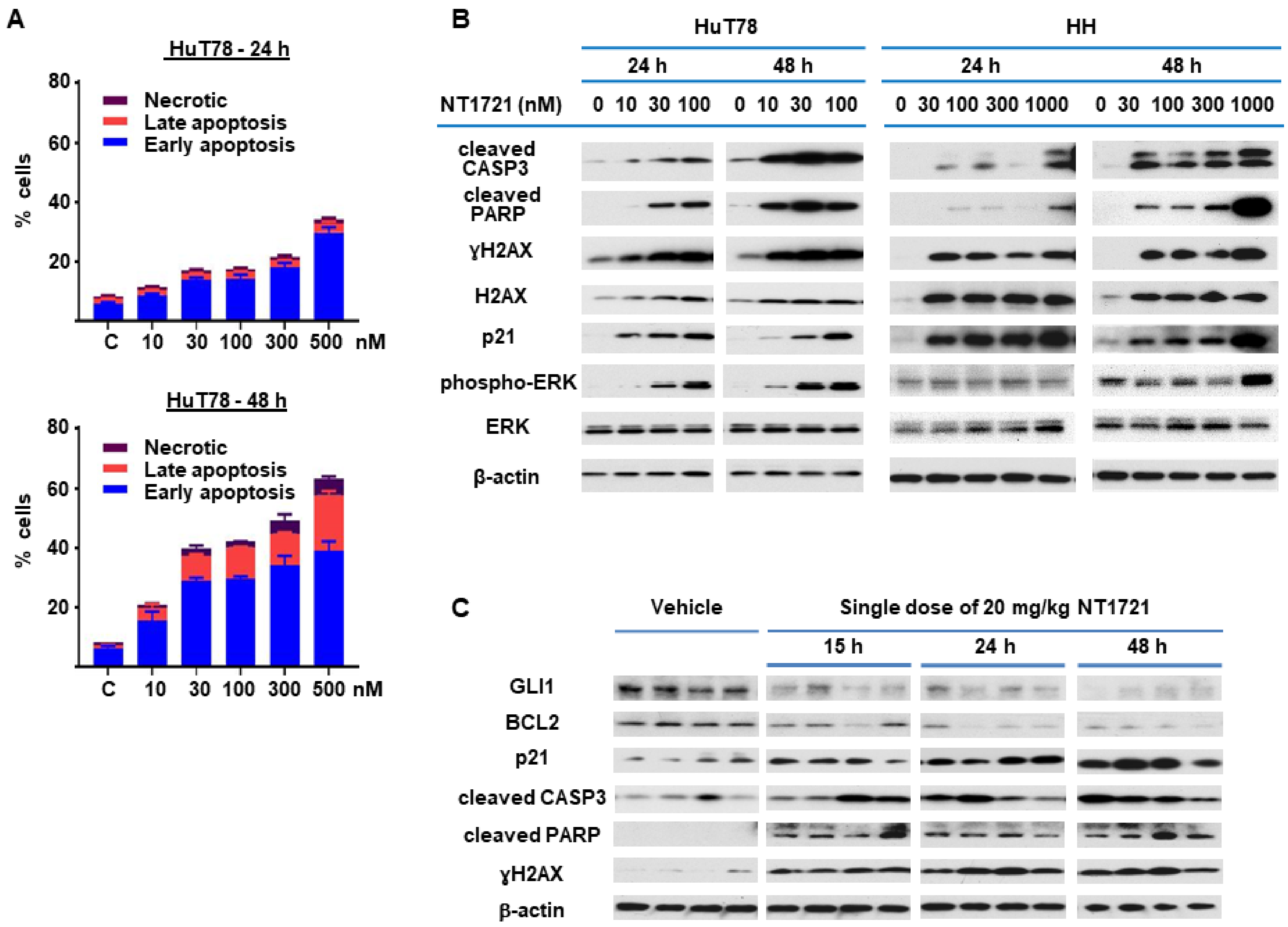
3.3. NT1721 Induced Apoptosis in CTCL In Vitro and In Vivo
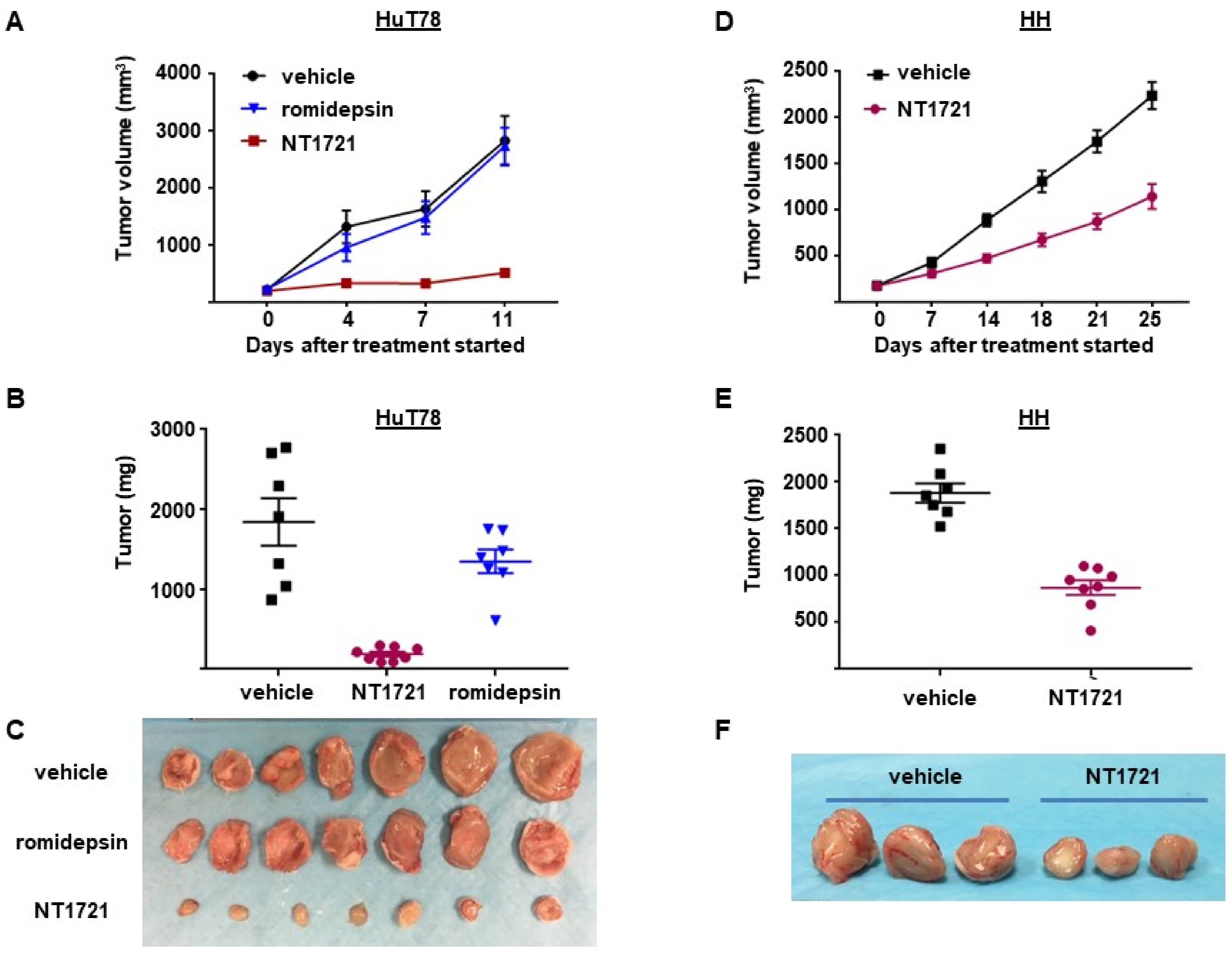
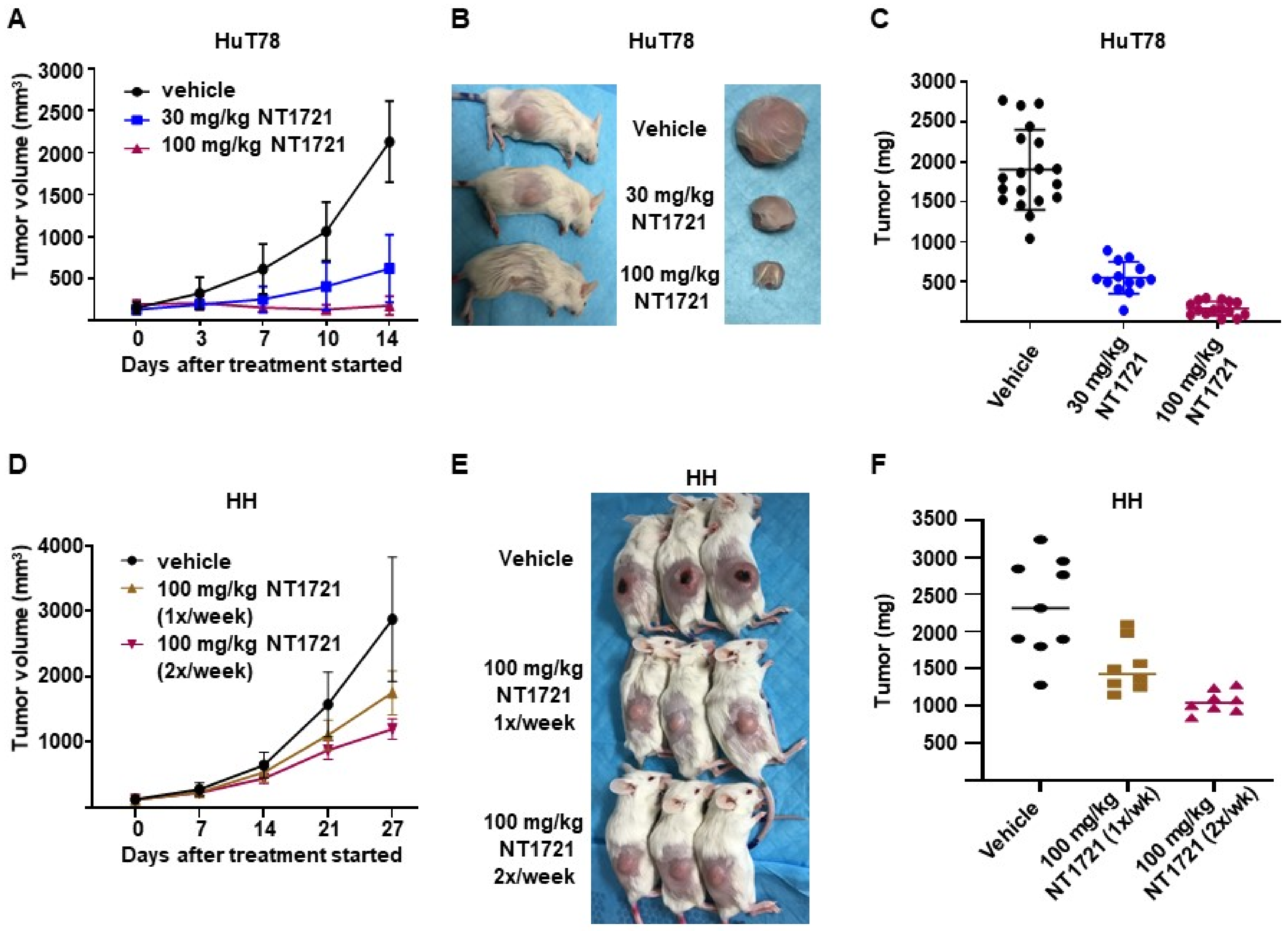
3.4. NT1721 Suppressed Tumor Growth Significantly Better Than Romidepsin in CTCL Mouse Models
3.5. DMSO-Free Formulation of NT1721 Potently Decreased CTCL Tumor Growth
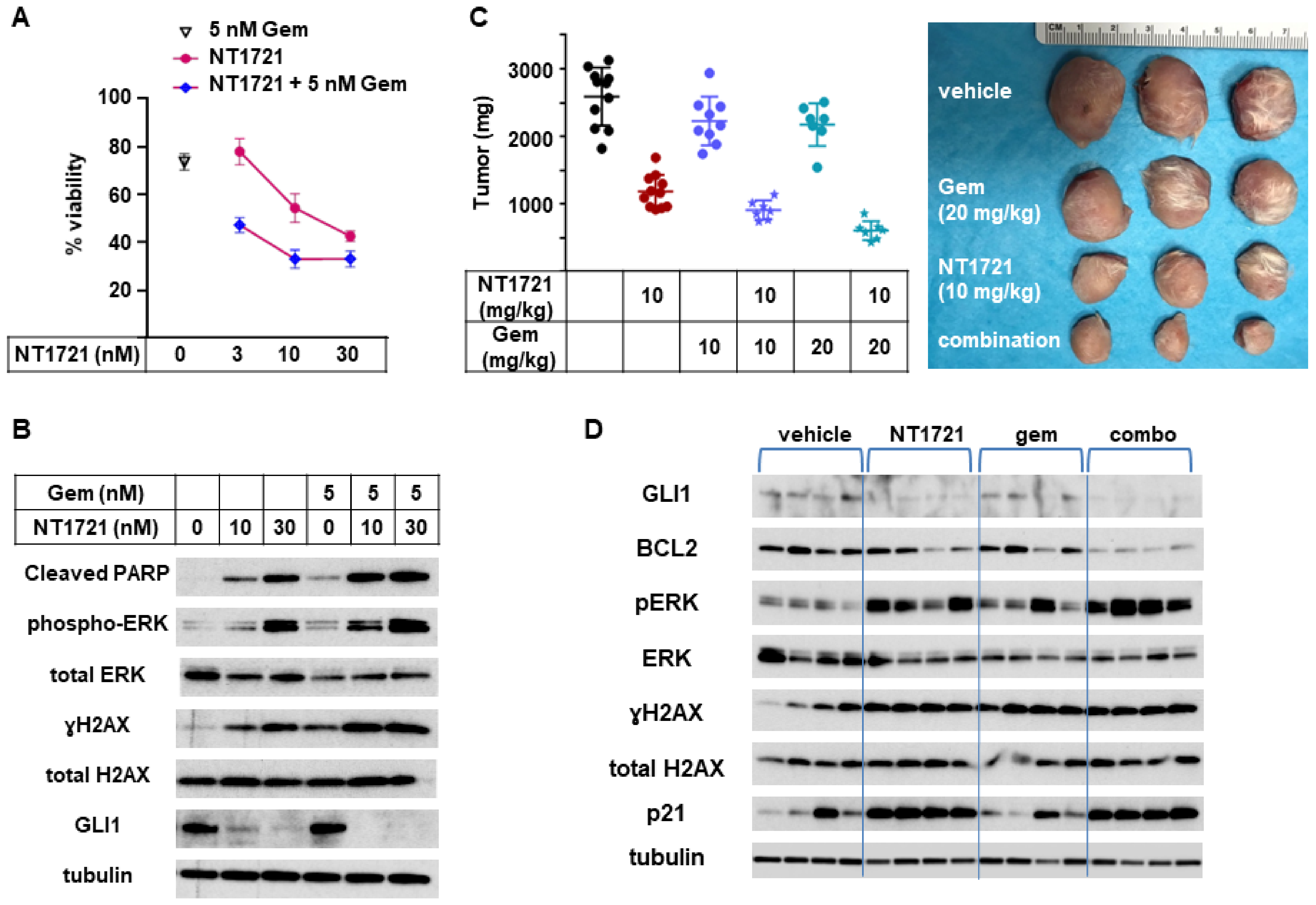
3.6. In Vivo Combinations of NT1721 and Gemcitabine Suppressed CTCL Tumor Growth Better Than the Single Agents
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bradford, P.T.; Devesa, S.S.; Anderson, W.F.; Toro, J.R. Cutaneous lymphoma incidence patterns in the United States: A population-based study of 3884 cases. Blood 2009, 113, 5064–5073. [Google Scholar] [CrossRef] [PubMed]
- Bagherani, N.; Smoller, B.R. An overview of cutaneous T cell lymphomas. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Criscione, V.D.; Weinstock, M.A. Incidence of cutaneous T-cell lymphoma in the United States, 1973–2002. Arch. Dermatol. 2007, 143, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Argnani, L.; Broccoli, A.; Zinzani, P.L. Cutaneous T-cell lymphomas: Focusing on novel agents in relapsed and refractory disease. Cancer Treat. Rev. 2017, 61, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K. New Therapies and Immunological Findings in Cutaneous T-Cell Lymphoma. Front. Oncol. 2018, 8, 198. [Google Scholar] [CrossRef]
- Dobos, G.; Pohrt, A.; Ram-Wolff, C.; Lebbe, C.; Bouaziz, J.D.; Battistella, M.; Bagot, M.; de Masson, A. Epidemiology of Cutaneous T-Cell Lymphomas: A Systematic Review and Meta-Analysis of 16,953 Patients. Cancers 2020, 12, 2921. [Google Scholar] [CrossRef]
- Kim, S.R.; Lewis, J.M.; Cyrenne, B.M.; Monico, P.F.; Mirza, F.N.; Carlson, K.R.; Foss, F.M.; Girardi, M. BET inhibition in advanced cutaneous T cell lymphoma is synergistically potentiated by BCL2 inhibition or HDAC inhibition. Oncotarget 2018, 9, 29193–29207. [Google Scholar] [CrossRef]
- Van Doorn, R.; van Kester, M.S.; Dijkman, R.; Vermeer, M.H.; Mulder, A.A.; Szuhai, K.; Knijnenburg, J.; Boer, J.M.; Willemze, R.; Tensen, C.P. Oncogenomic analysis of mycosis fungoides reveals major differences with Sezary syndrome. Blood 2009, 113, 127–136. [Google Scholar] [CrossRef]
- Campbell, J.J.; Clark, R.A.; Watanabe, R.; Kupper, T.S. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: A biologic rationale for their distinct clinical behaviors. Blood 2010, 116, 767–771. [Google Scholar] [CrossRef]
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef]
- Dulmage, B.O.; Geskin, L.J. Lessons learned from gene expression profiling of cutaneous T-cell lymphoma. Br. J. Dermatol. 2013, 169, 1188–1197. [Google Scholar] [CrossRef]
- Kremer, M.; Sliva, K.; Klemke, C.D.; Schnierle, B.S. Cutaneous T-cell lymphoma cells are sensitive to rapamycin. Exp. Dermatol. 2010, 19, 800–805. [Google Scholar] [CrossRef]
- Kim, E.J.; Hess, S.; Richardson, S.K.; Newton, S.; Showe, L.C.; Benoit, B.M.; Ubriani, R.; Vittorio, C.C.; Junkins-Hopkins, J.M.; Wysocka, M.; et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J. Clin. Investig. 2005, 115, 798–812. [Google Scholar] [CrossRef]
- Choi, J.; Goh, G.; Walradt, T.; Hong, B.S.; Bunick, C.G.; Chen, K.; Bjornson, R.D.; Maman, Y.; Wang, T.; Tordoff, J.; et al. Genomic landscape of cutaneous T cell lymphoma. Nat. Genet. 2015, 47, 1011–1019. [Google Scholar] [CrossRef]
- Janiga, J.; Kentley, J.; Nabhan, C.; Abdulla, F. Current systemic therapeutic options for advanced mycosis fungoides and Sezary syndrome. Leuk. Lymphoma 2018, 59, 562–577. [Google Scholar] [CrossRef]
- Rozati, S.; Cheng, P.F.; Widmer, D.S.; Fujii, K.; Levesque, M.P.; Dummer, R. Romidepsin and Azacitidine Synergize in their Epigenetic Modulatory Effects to Induce Apoptosis in CTCL. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 2020–2031. [Google Scholar] [CrossRef]
- Froehlich, T.C.; Muller-Decker, K.; Braun, J.D.; Albrecht, T.; Schroeder, A.; Gulow, K.; Goerdt, S.; Krammer, P.H.; Nicolay, J.P. Combined inhibition of Bcl-2 and NFkappaB synergistically induces cell death in cutaneous T-cell lymphoma. Blood 2019, 134, 445–455. [Google Scholar] [CrossRef]
- Damsky, W.E.; Choi, J. Genetics of Cutaneous T Cell Lymphoma: From Bench to Bedside. Curr. Treat. Options Oncol. 2016, 17, 33. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Tetzlaff, M.T.; Thibault, P.; Gangar, P.; Moreau, L.; Watters, A.K.; Netchiporouk, E.; Pehr, K.; Prieto, V.G.; Rahme, E.; et al. Gene expression analysis in Cutaneous T-Cell Lymphomas (CTCL) highlights disease heterogeneity and potential diagnostic and prognostic indicators. Oncoimmunology 2017, 6, e1306618. [Google Scholar] [CrossRef]
- Fanok, M.H.; Sun, A.; Fogli, L.K.; Narendran, V.; Eckstein, M.; Kannan, K.; Dolgalev, I.; Lazaris, C.; Heguy, A.; Laird, M.E.; et al. Role of Dysregulated Cytokine Signaling and Bacterial Triggers in the Pathogenesis of Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2018, 138, 1116–1125. [Google Scholar] [CrossRef]
- Hwang, S.T.; Janik, J.E.; Jaffe, E.S.; Wilson, W.H. Mycosis fungoides and Sezary syndrome. Lancet 2008, 371, 945–957. [Google Scholar] [CrossRef]
- Wu, J.; Nihal, M.; Siddiqui, J.; Vonderheid, E.C.; Wood, G.S. Low FAS/CD95 expression by CTCL correlates with reduced sensitivity to apoptosis that can be restored by FAS upregulation. J. Investig. Dermatol. 2009, 129, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Mondejar, R.; Garcia-Diaz, N.; Cereceda, L.; Leon, A.; Montes, S.; Duran Vian, C.; Perez Paredes, M.G.; Gonzalez-Moran, A.; Alegre de Miguel, V.; et al. Advanced-stage mycosis fungoides: Role of the signal transducer and activator of transcription 3, nuclear factor-kappaB and nuclear factor of activated T cells pathways. Br. J. Dermatol. 2020, 182, 147–155. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, R.C.; Jones, C.L.; Tosi, I.; Caesar, J.A.; Whittaker, S.J.; Mitchell, T.J. Constitutive activation of STAT3 in Sezary syndrome is independent of SHP-1. Leukemia 2012, 26, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Sommer, V.H.; Clemmensen, O.J.; Nielsen, O.; Wasik, M.; Lovato, P.; Brender, C.; Eriksen, K.W.; Woetmann, A.; Kaestel, C.G.; Nissen, M.H.; et al. In vivo activation of STAT3 in cutaneous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3. Leukemia 2004, 18, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Seffens, A.; Herrera, A.; Tegla, C.; Buus, T.B.; Hymes, K.B.; Odum, N.; Geskin, L.J.; Koralov, S.B. STAT3 Dysregulation in Mature T and NK Cell Lymphomas. Cancers 2019, 11, 1711. [Google Scholar] [CrossRef] [PubMed]
- Kowolik, C.M.; Lin, M.; Xie, J.; Overman, L.E.; Horne, D.A. Attenuation of hedgehog/GLI signaling by NT1721 extends survival in pancreatic cancer. J. Exp. Clin. Cancer Res. CR 2019, 38, 431. [Google Scholar] [CrossRef]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef]
- Doheny, D.; Manore, S.G.; Wong, G.L.; Lo, H.W. Hedgehog Signaling and Truncated GLI1 in Cancer. Cells 2020, 9, 2114. [Google Scholar] [CrossRef]
- Duvic, M.; Talpur, R.; Wen, S.; Kurzrock, R.; David, C.L.; Apisarnthanarax, N. Phase II evaluation of gemcitabine monotherapy for cutaneous T-cell lymphoma. Clin. Lymphoma Myeloma 2006, 7, 51–58. [Google Scholar] [CrossRef]
- Duvic, M.; Bates, S.E.; Piekarz, R.; Eisch, R.; Kim, Y.H.; Lerner, A.; Robak, T.; Samtsov, A.; Becker, J.C.; McCulloch, W.; et al. Responses to romidepsin in patients with cutaneous T-cell lymphoma and prior treatment with systemic chemotherapy. Leuk. Lymphoma 2018, 59, 880–887. [Google Scholar] [CrossRef]
- Baumann, M.; Dieskau, A.P.; Loertscher, B.M.; Walton, M.C.; Nam, S.; Xie, J.; Horne, D.; Overman, L.E. Tricyclic Analogues of Epidithiodioxopiperazine Alkaloids with Promising and Antitumor Activity. Chem. Sci. 2015, 6, 4451–4457. [Google Scholar] [CrossRef]
- Geng, L.; Lu, K.; Li, P.; Li, X.; Zhou, X.; Li, Y.; Wang, X. GLI1 inhibitor GANT61 exhibits antitumor efficacy in T-cell lymphoma cells through down-regulation of p-STAT3 and SOCS3. Oncotarget 2017, 8, 48701–48710. [Google Scholar] [CrossRef]
- Chen, J.; Fiskus, W.; Eaton, K.; Fernandez, P.; Wang, Y.; Rao, R.; Lee, P.; Joshi, R.; Yang, Y.; Kolhe, R.; et al. Cotreatment with BCL-2 antagonist sensitizes cutaneous T-cell lymphoma to lethal action of HDAC7-Nur77-based mechanism. Blood 2009, 113, 4038–4048. [Google Scholar] [CrossRef]
- Liu, Y.; Tseng, M.; Perdreau, S.A.; Rossi, F.; Antonescu, C.; Besmer, P.; Fletcher, J.A.; Duensing, S.; Duensing, A. Histone H2AX is a mediator of gastrointestinal stromal tumor cell apoptosis following treatment with imatinib mesylate. Cancer Res. 2007, 67, 2685–2692. [Google Scholar] [CrossRef]
- Liu, Y.; Parry, J.A.; Chin, A.; Duensing, S.; Duensing, A. Soluble histone H2AX is induced by DNA replication stress and sensitizes cells to undergo apoptosis. Mol. Cancer 2008, 7, 61. [Google Scholar] [CrossRef]
- Tang, D.; Wu, D.; Hirao, A.; Lahti, J.M.; Liu, L.; Mazza, B.; Kidd, V.J.; Mak, T.W.; Ingram, A.J. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J. Biol. Chem. 2002, 277, 12710–12717. [Google Scholar] [CrossRef]
- Sun, W.J.; Huang, H.; He, B.; Hu, D.H.; Li, P.H.; Yu, Y.J.; Zhou, X.H.; Lv, Z.; Zhou, L.; Hu, T.Y.; et al. Romidepsin induces G2/M phase arrest via Erk/cdc25C/cdc2/cyclinB pathway and apoptosis induction through JNK/c-Jun/caspase3 pathway in hepatocellular carcinoma cells. Biochem. Pharmacol. 2017, 127, 90–100. [Google Scholar] [CrossRef]
- Prince, H.M.; Querfeld, C. Integrating novel systemic therapies for the treatment of mycosis fungoides and Sezary syndrome. Best Pract. Res. Clin. Haematol. 2018, 31, 322–335. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Lindahl, L.M.; Mongan, N.P.; Wasik, M.A.; Litvinov, I.V.; Iversen, L.; Langhoff, E.; Woetmann, A.; Odum, N. Malignant inflammation in cutaneous T-cell lymphoma-a hostile takeover. Semin. Immunopathol. 2017, 39, 269–282. [Google Scholar] [CrossRef]
- Singh, R.R.; Cho-Vega, J.H.; Davuluri, Y.; Ma, S.; Kasbidi, F.; Milito, C.; Lennon, P.A.; Drakos, E.; Medeiros, L.J.; Luthra, R.; et al. Sonic hedgehog signaling pathway is activated in ALK-positive anaplastic large cell lymphoma. Cancer Res. 2009, 69, 2550–2558. [Google Scholar] [CrossRef]
- Gonzalez-Gugel, E.; Villa-Morales, M.; Santos, J.; Bueno, M.J.; Malumbres, M.; Rodriguez-Pinilla, S.M.; Piris, M.A.; Fernandez-Piqueras, J. Down-regulation of specific miRNAs enhances the expression of the gene Smoothened and contributes to T-cell lymphoblastic lymphoma development. Carcinogenesis 2013, 34, 902–908. [Google Scholar] [CrossRef]
- Liu, S.; Li, C.; Xin, P.; Zheng, Y.; Peng, Q.; Xu, Y.; Luo, Y.; Wu, Y.; Zhu, X. Sonidegib, a Smoothened Inhibitor, Promotes Apoptosis and Suppresses Proliferation of Natural Killer/T-Cell Lymphoma. Med Sci. Monit. Int. Med J. Exp. Clin. Res. 2019, 25, 8579–8586. [Google Scholar] [CrossRef]
- Van Kester, M.S.; Out-Luiting, J.J.; von dem Borne, P.A.; Willemze, R.; Tensen, C.P.; Vermeer, M.H. Cucurbitacin I inhibits Stat3 and induces apoptosis in Sezary cells. J. Investig. Dermatol. 2008, 128, 1691–1695. [Google Scholar] [CrossRef] [PubMed]
- Cyrenne, B.M.; Lewis, J.M.; Weed, J.G.; Carlson, K.R.; Mirza, F.N.; Foss, F.M.; Girardi, M. Synergy of BCL2 and histone deacetylase inhibition against leukemic cells from cutaneous T-cell lymphoma patients. Blood 2017, 130, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Yumeen, S.; Mirza, F.N.; Lewis, J.M.; King, A.L.O.; Kim, S.R.; Carlson, K.R.; Umlauf, S.R.; Surovtseva, Y.V.; Foss, F.M.; Girardi, M. JAK inhibition synergistically potentiates BCL2, BET, HDAC, and proteasome inhibition in advanced CTCL. Blood Adv. 2020, 4, 2213–2226. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, L.S.; Toombs, J.; Kuo, Y.C.; Piazza, J.T.; Tuladhar, R.; Barrett, Q.; Fan, C.W.; Zhang, X.; Walensky, L.D.; et al. Extra-mitochondrial prosurvival BCL-2 proteins regulate gene transcription by inhibiting the SUFU tumour suppressor. Nat. Cell Biol. 2017, 19, 1226–1236. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, M.; Kowolik, C.M.; Xie, J.; Yadav, S.; Overman, L.E.; Horne, D.A. Potent Anticancer Effects of Epidithiodiketopiperazine NT1721 in Cutaneous T Cell Lymphoma. Cancers 2021, 13, 3367. https://doi.org/10.3390/cancers13133367
Lin M, Kowolik CM, Xie J, Yadav S, Overman LE, Horne DA. Potent Anticancer Effects of Epidithiodiketopiperazine NT1721 in Cutaneous T Cell Lymphoma. Cancers. 2021; 13(13):3367. https://doi.org/10.3390/cancers13133367
Chicago/Turabian StyleLin, Min, Claudia M. Kowolik, Jun Xie, Sushma Yadav, Larry E. Overman, and David A. Horne. 2021. "Potent Anticancer Effects of Epidithiodiketopiperazine NT1721 in Cutaneous T Cell Lymphoma" Cancers 13, no. 13: 3367. https://doi.org/10.3390/cancers13133367