
Single Cell Genetic Profiling of Tumors of Breast Cancer Patients Aged 50 Years and Older Reveals Enormous Intratumor Heterogeneity Independent of Individual Prognosis

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
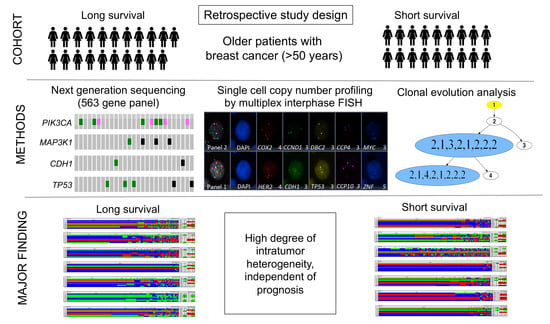
1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Preparation of Cytospins from Archival FFPE Specimens
2.3. Multiplex Interphase Fluorescence in Situ Hybridization (miFISH)
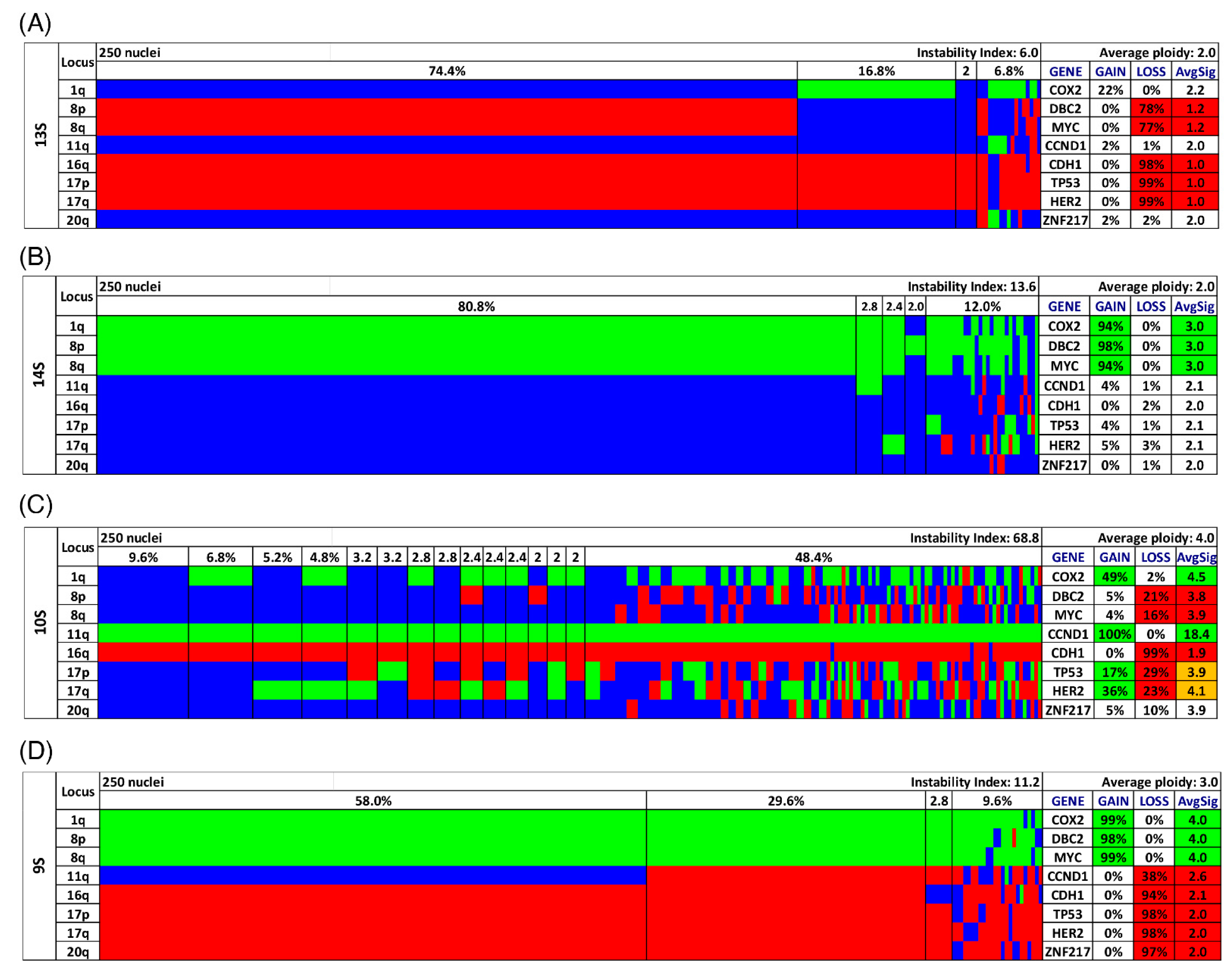
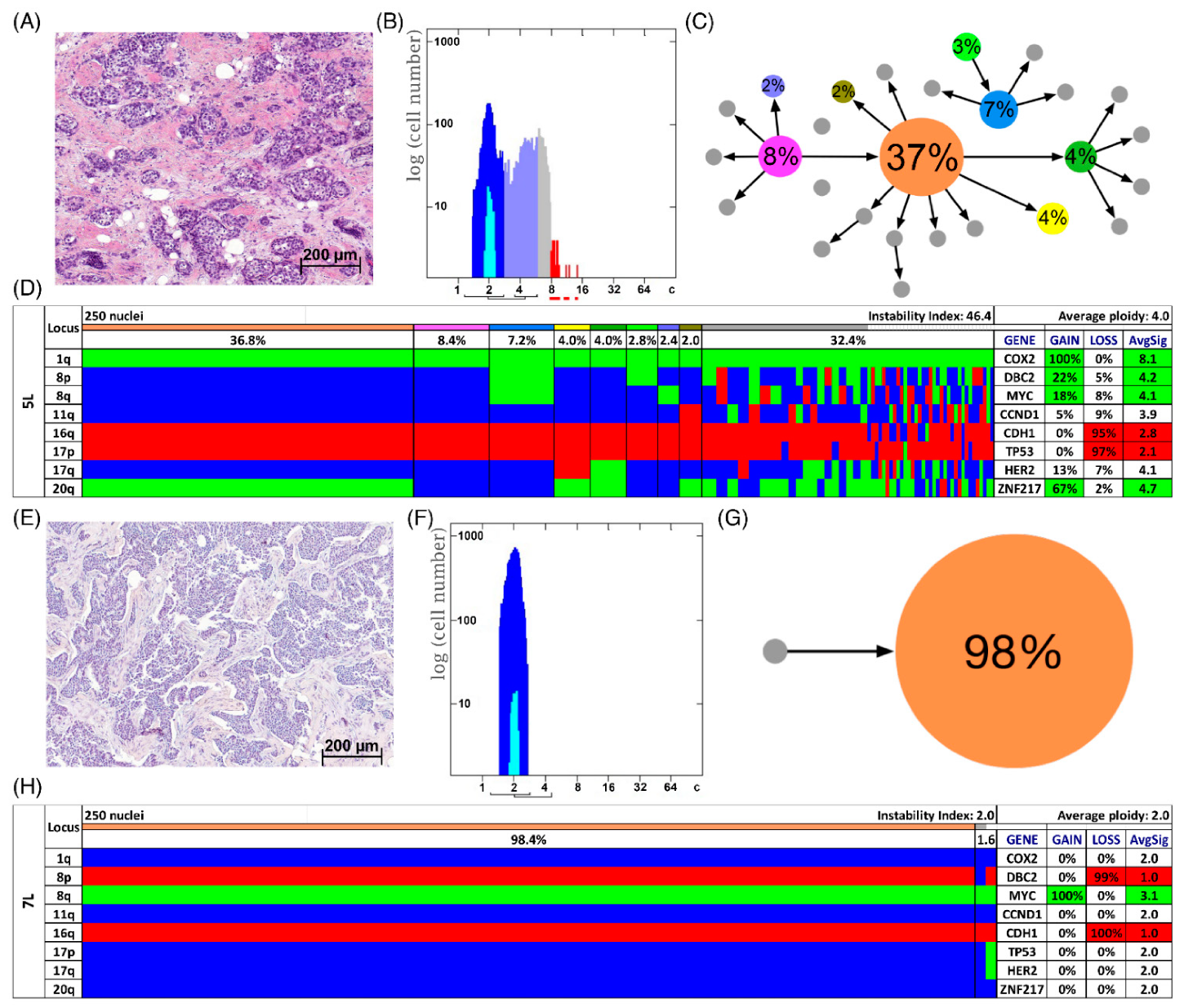
2.4. Determining Clonal Signal Patterns, Gain and Loss Patterns, Ploidy and Instability Index
2.5. Quantitative Measurement of the Nuclear DNA Content by Image Cytometry
2.6. Clonal Evolution in Tumors Assessed by Phylogenetic Tree Modelling
2.7. Targeted Next Generation Sequencing, Sequencing Data Processing, and Analyses
2.8. Statistics
3. Results
3.1. Clinicopathologic Characteristics
3.2. Landscape of Gene Mutations
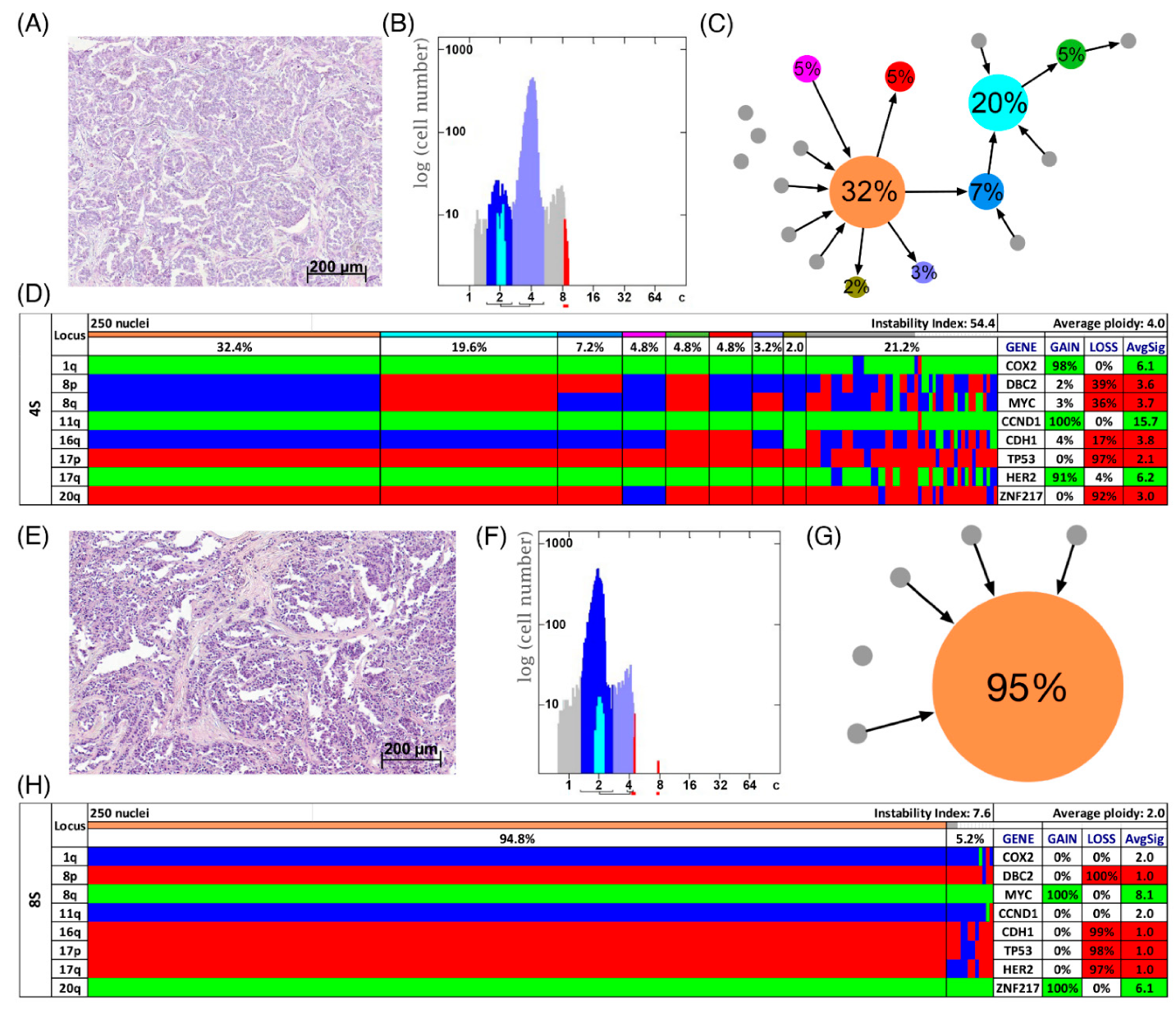
3.3. Analysis of CNAs and ITH by miFISH
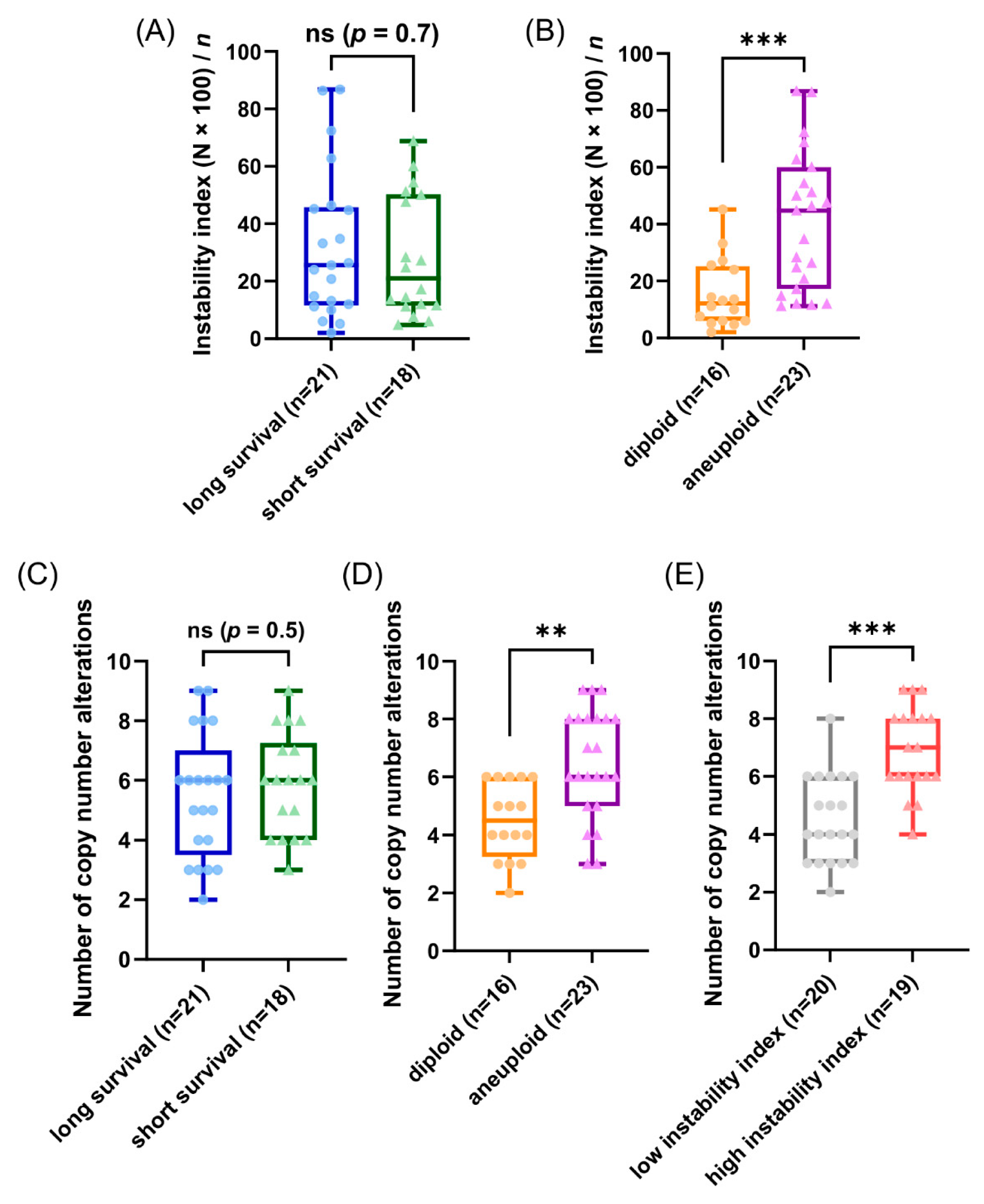
3.4. Genetic Characteristics of Subgroups Distinct by Survival Time, Ploidy and Instability Index
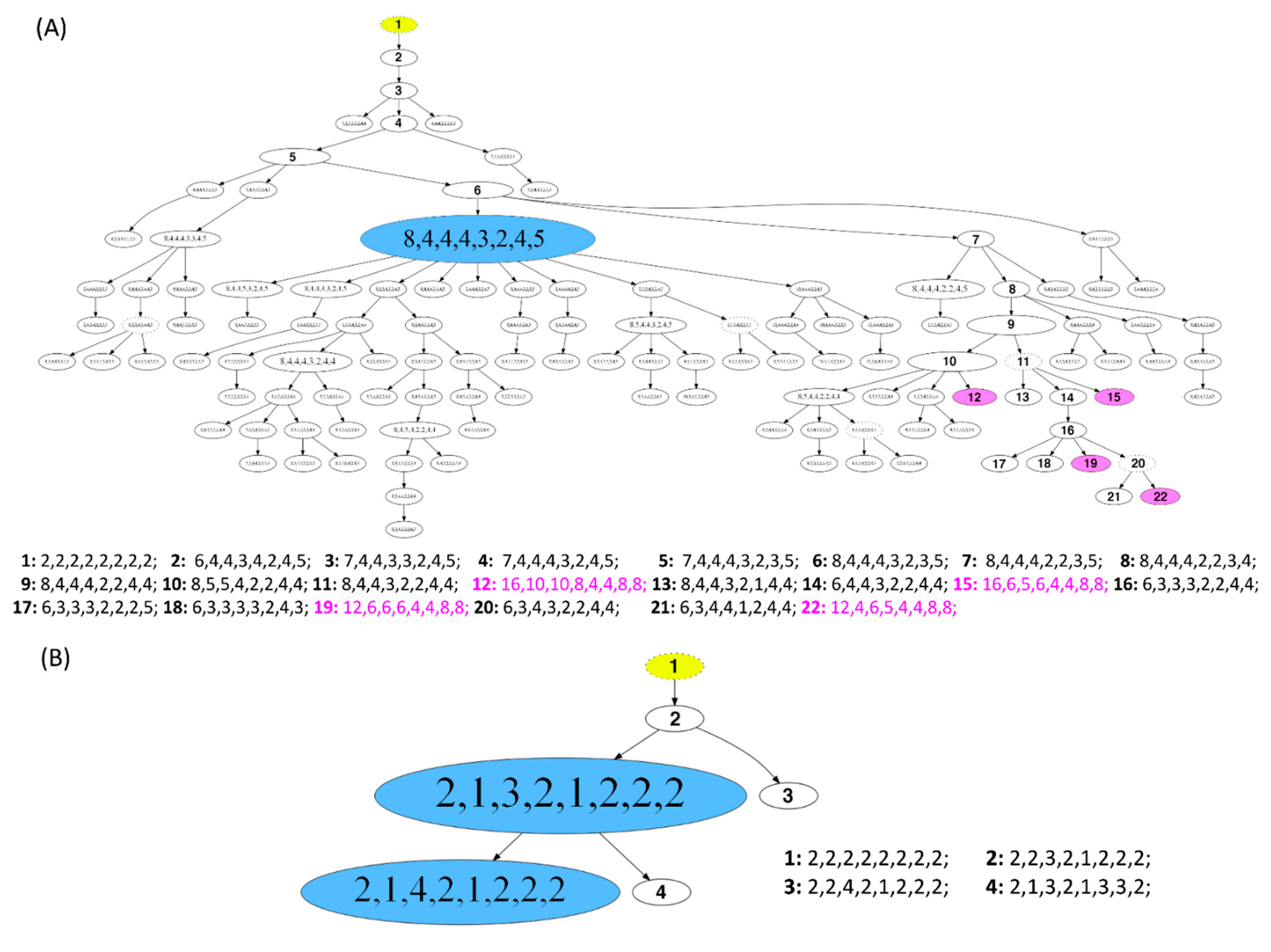
3.5. Phylogenetic Analysis by FISHtree Modelling
3.6. Mutual Exclusivity und Co-Occurrence Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Surveillance, Epidemiology, and End Results (SEER) Program. Cancer Stat Facts: Female Breast Cancer. Available online: https://seer.cancer.gov/statfacts/html/breast.html (accessed on 5 April 2020).
- U.S. Department of Health and Human Services. 2017 Profile of Older Americans. Available online: https://acl.gov/sites/default/files/Aging%20and%20Disability%20in%20America/2017OlderAmericansProfile.pdf (accessed on 21 April 2021).
- Smith, B.; Smith, G.L.; Hurria, A.; Hortobagyi, G.N.; Buchholz, T.A. Future of Cancer Incidence in the United States: Burdens Upon an Aging, Changing Nation. J. Clin. Oncol. 2009, 27, 2758–2765. [Google Scholar] [CrossRef]
- Hurria, A.; Levit, L.A.; Dale, W.; Mohile, S.G.; Muss, H.B.; Fehrenbacher, L.; Magnuson, A.; Lichtman, S.; Bruinooge, S.S.; Soto-Perez-De-Celis, E.; et al. Improving the Evidence Base for Treating Older Adults with Cancer: American Society of Clinical Oncology Statement. J. Clin. Oncol. 2015, 33, 3826–3833. [Google Scholar] [CrossRef] [PubMed]
- Dale, W.; Mohile, S.G.; Eldadah, B.A.; Trimble, E.L.; Schilsky, R.L.; Cohen, H.J.; Muss, H.B.; Schmader, K.E.; Ferrell, B.; Extermann, M.; et al. Biological, Clinical, and Psychosocial Correlates at the Interface of Cancer and Aging Research. J Natl. Cancer Inst. 2012, 104, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Martelotto, L.G.; Ng, C.K.Y.; Piscuoglio, S.; Weigelt, B.; Reis-Filho, J.S. Breast cancer intra-tumor heterogeneity. Breast Cancer Res. 2014, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Beca, F.; Polyak, K. Intratumor Heterogeneity in Breast Cancer. Adv. Exp. Med. Biol. 2016, 882, 169–189. [Google Scholar] [CrossRef]
- Oltmann, J.; Heselmeyer-Haddad, K.; Hernandez, L.S.; Meyer, R.; Torres, I.; Hu, Y.; Doberstein, N.; Killian, J.K.; Petersen, D.; Zhu, Y.J.; et al. Aneuploidy, TP53 mutation, and amplification of MYC correlate with increased intratumor heterogeneity and poor prognosis of breast cancer patients. Genes Chromosomes Cancer 2017, 57, 165–175. [Google Scholar] [CrossRef]
- Koçak, A.; Heselmeyer-Haddad, K.; Lischka, A.; Hirsch, D.; Fiedler, D.; Hu, Y.; Doberstein, N.; Torres, I.; Chen, W.-D.; Gertz, E.M.; et al. High Levels of Chromosomal Copy Number Alterations and TP53 Mutations Correlate with Poor Outcome in Younger Breast Cancer Patients. Am. J. Pathol. 2020, 190, 1643–1656. [Google Scholar] [CrossRef]
- Heselmeyer-Haddad, K.; Garcia, L.Y.B.; Bradley, A.; Ortiz-Melendez, C.; Lee, W.-J.; Christensen, R.; Prindiville, S.A.; Calzone, K.A.; Soballe, P.W.; Hu, Y.; et al. Single-Cell Genetic Analysis of Ductal Carcinoma in Situ and Invasive Breast Cancer Reveals Enormous Tumor Heterogeneity yet Conserved Genomic Imbalances and Gain of MYC during Progression. Am. J. Pathol. 2012, 181, 1807–1822. [Google Scholar] [CrossRef] [Green Version]
- Martelotto, L.G.; Baslan, T.; Kendall, J.; Geyer, F.C.; Burke, K.A.; Spraggon, L.; Piscuoglio, S.; Chadalavada, K.; Nanjangud, G.; Ng, C.K.Y.; et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nat. Med. 2017, 23, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef] [Green Version]
- Janiszewska, M.; Liu, L.; Almendro, V.; Kuang, Y.; Paweletz, C.; Sakr, R.A.; Weigelt, B.; Hanker, A.B.; Chandarlapaty, S.; King, T.A.; et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat. Genet. 2015, 47, 1212–1219. [Google Scholar] [CrossRef] [Green Version]
- Paik, S.; Tang, G.; Shak, S.; Kim, C.; Baker, J.; Kim, W.; Cronin, M.; Baehner, F.L.; Watson, D.; Bryant, J.; et al. Gene Expression and Benefit of Chemotherapy in Women with Node-Negative, Estrogen Receptor–Positive Breast Cancer. J. Clin. Oncol. 2006, 24, 3726–3734. [Google Scholar] [CrossRef]
- Van’T Veer, L.J.; Dai, H.; Van De Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; Van Der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Filipits, M.; Rudas, M.; Jakesz, R.; Dubsky, P.; Fitzal, F.; Singer, C.F.; Dietze, O.; Greil, R.; Jelen, A.; Sevelda, P.; et al. A New Molecular Predictor of Distant Recurrence in ER-Positive, HER2-Negative Breast Cancer Adds Independent Information to Conventional Clinical Risk Factors. Clin. Cancer Res. 2011, 17, 6012–6020. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.J.; O’Donovan, N.; McDermott, E.; Crown, J. Validated biomarkers: The key to precision treatment in patients with breast cancer. Breast 2016, 29, 192–201. [Google Scholar] [CrossRef]
- Auer, G.; Eriksson, E.; Azavedo, E.; Caspersson, T.; Wallgren, A. Prognostic significance of nuclear DNA content in mammary adenocarcinomas in humans. Cancer Res. 1984, 44, 394–396. [Google Scholar]
- Lischka, A.; Doberstein, N.; Freitag-Wolf, S.; Koçak, A.; Gemoll, T.; Heselmeyer-Haddad, K.; Ried, T.; Auer, G.; Habermann, J.K. Genome Instability Profiles Predict Disease Outcome in a Cohort of 4,003 Patients with Breast Cancer. Clin. Cancer Res. 2020, 26, 4606–4615. [Google Scholar] [CrossRef]
- Kronenwett, U.; Ploner, A.; Zetterberg, A.; Bergh, J.; Hall, P.; Auer, G.; Pawitan, Y. Genomic Instability and Prognosis in Breast Carcinomas. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1630–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Ruan, S.; Zhao, X.; Shi, X.; Teng, H.; Zhong, J.; You, M.; Xia, K.; Sun, Z.; Mao, F. OncoVar: An integrated database and analysis platform for oncogenic driver variants in cancers. Nucleic Acids Res. 2021, 49, D1289–D1301. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.H.; Schwartz, J.N.; Hagerty, K.L.; Allred, D.C.; Cote, R.J.; Dowsett, M.; Fitzgibbons, P.L.; Hanna, W.M.; Langer, A.; et al. American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer. Arch. Pathol. Lab. Med. 2007, 131, 18–43. [Google Scholar] [CrossRef]
- Hammond, M.E.H.; Hayes, D.F.; Wolff, A.; Mangu, P.B.; Temin, S. American Society of Clinical Oncology/College of American Pathologists Guideline Recommendations for Immunohistochemical Testing of Estrogen and Progesterone Receptors in Breast Cancer. J. Oncol. Pract. 2010, 6, 195–197. [Google Scholar] [CrossRef] [Green Version]
- Dowsett, M.; Nielsen, T.O.; A’Hern, R.; Bartlett, J.; Coombes, R.C.; Cuzick, J.; Ellis, M.; Henry, N.L.; Hugh, J.C.; Lively, T.; et al. Assessment of Ki67 in Breast Cancer: Recommendations from the International Ki67 in Breast Cancer Working Group. J. Natl. Cancer Inst. 2011, 103, 1656–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J.; Albain, K.S.; Andre, F.; Bergh, J.; et al. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef] [PubMed]
- Tirkkonen, M.; Tanner, M.; Karhu, R.; Kallioniemi, A.; Isola, J.; Kallioniemi, O.P. Molecular Cytogenetics of Primary Breast Cancer by CGH. Genes Chromosomes Cancer 1998, 21, 177–184. [Google Scholar] [CrossRef]
- Ried, T.; Just, K.E.; Holtgreve-Grez, H.; du Manoir, S.; Speicher, M.; Schröck, E.; Latham, C.; Blegen, H.; Zetterberg, A.; Cremer, T. Comparative genomic hybridization of formalin-fixed, paraffin-embedded breast tumors reveals different patterns of chromosomal gains and losses in fibroadenomas and diploid and aneuploid carcinomas. Cancer Res. 1995, 55, 5415–5423. [Google Scholar]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Auer, G.U.; Caspersson, T.O.; Wallgren, A.S. DNA content and survival in mammary carcinoma. Anal. Quant. Cytol. 1980, 2, 161–165. [Google Scholar]
- Gertz, E.M.; Chowdhury, S.A.; Lee, W.-J.; Wangsa, D.; Heselmeyer-Haddad, K.; Ried, T.; Schwartz, R.; Schäffer, A.A. FISHtrees 3.0: Tumor Phylogenetics Using a Ploidy Probe. PLoS ONE 2016, 11, e0158569. [Google Scholar] [CrossRef]
- Killian, J.K.; Miettinen, M.; Walker, R.L.; Wang, Y.; Zhu, Y.J.; Waterfall, J.; Noyes, N.; Retnakumar, P.; Yang, Z.; Smith, W.I.; et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci. Transl. Med. 2014, 6, 268ra177. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.; Cummings, B.; et al. The ExAC browser: Displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriello, G.; Cerami, E.; Sander, C.; Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2011, 22, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Cerami, E.; Aksoy, B.A.; Sander, C.; Schultz, N. Using MEMo to Discover Mutual Exclusivity Modules in Cancer. Curr. Protoc. Bioinform. 2013, 41, 8.17.1–8.17.12. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, D.; Wangsa, D.; Zhu, Y.J.; Hu, Y.; Edelman, D.C.; Meltzer, P.S.; Heselmeyer-Haddad, K.; Ott, C.; Kienle, P.; Galata, C.; et al. Dynamics of Genome Alterations in Crohn’s Disease–Associated Colorectal Carcinogenesis. Clin. Cancer Res. 2018, 24, 4997–5011. [Google Scholar] [CrossRef] [Green Version]
- Vorkas, P.A.; Poumpouridou, N.; Agelaki, S.; Kroupis, C.; Georgoulias, V.; Lianidou, E.S. PIK3CA Hotspot Mutation Scanning by a Novel and Highly Sensitive High-Resolution Small Amplicon Melting Analysis Method. J. Mol. Diagn. 2010, 12, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Rummukainen, J.; Kytölä, S.; Karhu, R.; Farnebo, F.; Larsson, C.; Isola, J.J. Aberrations of chromosome 8 in 16 breast cancer cell lines by comparative genomic hybridization, fluorescence in situ hybridization, and spectral karyotyping. Cancer Genet. Cytogenet. 2001, 126, 1–7. [Google Scholar] [CrossRef]
- Kallioniemi, A.; Kallioniemi, O.; Piper, J.; Tanner, M.; Stokke, T.; Chen, L.; Smith, H.S.; Pinkel, D.; Gray, J.W.; Waldman, F.M. Detection and mapping of amplified DNA sequences in breast cancer by comparative genomic hybridization. Proc. Natl. Acad. Sci. USA 1994, 91, 2156–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, F.; Johansson, B.; Mitelman, F. Isochromosomes in neoplasia. Genes, Chromosom. Cancer 1994, 10, 221–230. [Google Scholar] [CrossRef]
- Anbazhagan, R.; Fujii, H.; Gabrielson, E. Allelic loss of chromosomal arm 8p in breast cancer progression. Am. J. Pathol. 1998, 152, 815–819. [Google Scholar] [PubMed]
- Yarernko, M.L.; Recant, W.M.; Westbrook, C.A. Loss of heterozygosity from the short arm of chromosome 8 is an early event in breast cancers. Genes Chromosom. Cancer 1995, 13, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Riou, G.; Mathieu, M.-C.; Barrois, M.; Le Bihan, M.-L.; Ahomadegbe, J.-C.; Zvibel, I.; Brill, S.; Halpern, Z.; Moskovitz, S.; Yayon, A.; et al. c-erbB-2 (HER-2/neu) gene amplification is a better indicator of poor prognosis than protein over-expression in operable breast-cancer patients. Int. J. Cancer 2001, 95, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Gilcrease, M.Z.; Woodward, W.; Nicolas, M.M.; Corley, L.J.; Fuller, G.; Esteva, F.J.; Tucker, S.L.; Buchholz, T.A. Even Low-level HER2 Expression May be Associated with Worse Outcome in Node-positive Breast Cancer. Am. J. Surg. Pathol. 2009, 33, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Parikh, M.; Galkin, M.; Brunson, A.; Keegan, T.; Chew, H.K. Breast cancer-specific survival in patients with HER2-positive, node-negative T1a and T1b breast cancer. Cancer Treat. Res. Commun. 2018, 16, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurti, U.; Silverman, J.F. HER2 in Breast Cancer: A Review and Update. Adv. Anat. Pathol. 2014, 21, 100–107. [Google Scholar] [CrossRef]
- Asif, H.M.; Sultana, S.; Ahmed, S.; Akhtar, N.; Tariq, M. HER-2 Positive Breast Cancer—A Mini-Review. Asian Pac. J. Cancer Prev. 2016, 17, 1609–1615. [Google Scholar] [CrossRef] [Green Version]
- Roy, P.G.; Pratt, N.; Purdie, C.A.; Baker, L.; Ashfield, A.; Quinlan, P.; Thompson, A.M. High CCND1 amplification identifies a group of poor prognosis women with estrogen receptor positive breast cancer. Int. J. Cancer 2009, 127, 355–360. [Google Scholar] [CrossRef]
- Aaltonen, K.; Amini, R.-M.; Landberg, G.; Eerola, H.; Aittomäki, K.; Heikkilä, P.; Nevanlinna, H.; Blomqvist, C. Cyclin D1 expression is associated with poor prognostic features in estrogen receptor positive breast cancer. Breast Cancer Res. Treat. 2008, 113, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Habermann, J.K.; Doering, J.; Hautaniemi, S.; Roblick, U.J.; Bündgen, N.K.; Nicorici, D.; Kronenwett, U.; Rathnagiriswaran, S.; Mettu, R.K.R.; Ma, Y.; et al. The gene expression signature of genomic instability in breast cancer is an independent predictor of clinical outcome. Int. J. Cancer 2009, 124, 1552–1564. [Google Scholar] [CrossRef] [Green Version]
- Cornelisse, C.J.; Van De Velde, C.J.H.; Caspers, R.J.C.; Moolenaar, A.J.; Hermans, J. DNA ploidy and survival in breast cancer patients. Cytometry 1987, 8, 225–234. [Google Scholar] [CrossRef] [PubMed]
- van Jaarsveld, R.H.; Kops, G.J. Difference Makers: Chromosomal Instability versus Aneuploidy in Cancer. Trends Cancer 2016, 2, 561–571. [Google Scholar] [CrossRef]
- Potapova, T.A.; Zhu, J.; Li, R. Aneuploidy and chromosomal instability: A vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. 2013, 32, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Cook, K.R.; Vincent, L.; Hall, C.S.; Berry, J.A.; Multani, A.S.; Lucci, A. Cyclooxygenase-2 Induces Genomic Instability, BCL2 Expression, Doxorubicin Resistance, and Altered Cancer-Initiating Cell Phenotype in MCF7 Breast Cancer Cells. J. Surg. Res. 2008, 147, 240–246. [Google Scholar] [CrossRef]
- Rohrberg, J.; Van de Mark, D.; Amouzgar, M.; Lee, J.V.; Taileb, M.; Corella, A.; Kilinc, S.; Williams, J.; Jokisch, M.-L.; Camarda, R.; et al. MYC Dysregulates Mitosis, Revealing Cancer Vulnerabilities. Cell Rep. 2020, 30, 3368–3382.e7. [Google Scholar] [CrossRef] [Green Version]
- Casimiro, M.C.; Di Sante, G.; Crosariol, M.; Loro, E.; Dampier, W.; Ertel, A.; Yu, Z.; Saria, E.A.; Papanikolaou, A.; Li, Z.; et al. Kinase-independent role of cyclin D1 in chromosomal instability and mammary tumorigenesis. Oncotarget 2015, 6, 8525–8538. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Kim, H.J.; Jang, M.H.; Lee, S.; Ahn, S.; Park, S.Y. Centromere 17 copy number gain reflects chromosomal instability in breast cancer. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tanner, M.M.; Karhu, R.A.; Nupponen, N.N.; Borg, A.; Baldetorp, B.; Pejovic, T.; Fernö, M.; Killander, D.; Isola, J.J. Genetic Aberrations in Hypodiploid Breast Cancer: Frequent Loss of Chromosome 4 and Amplification of Cyclin D1 Oncogene. Am. J. Pathol. 1998, 153, 191–199. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nat. Cell Biol. 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.D.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.-B.; Brown, A.M.K.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.A.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Gertz, E.M.; Schäffer, A.A.; Fu, X.; Tao, Y.; Heselmeyer-Haddad, K.; Torres, I.; Shi, X.; Wu, K.; Li, G.; et al. Tumor Heterogeneity Assessed by Sequencing and Fluorescence in Situ Hybridization (FISH) Data. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Bader, A.G.; Vogt, P.K. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. USA 2005, 102, 802–807. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.X.; Polley, E.; Lipkowitz, S. New insights on PI3K/AKT pathway alterations and clinical outcomes in breast cancer. Cancer Treat. Rev. 2016, 45, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2010, 39, D945–D950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Jiang, Z.; Gao, Y.; Wang, L.; Chen, C.; Wang, X. TP53Mutations Promote Immunogenic Activity in Breast Cancer. J. Oncol. 2019, 2019, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Bertheau, P.; Lehmann-Che, J.; Varna, M.; Dumay, A.; Poirot, B.; Porcher, R.; Turpin, E.; Plassa, L.-F.; de Roquancourt, A.; Bourstyn, E.; et al. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast 2013, 22, S27–S29. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liegmann, A.-S.; Heselmeyer-Haddad, K.; Lischka, A.; Hirsch, D.; Chen, W.-D.; Torres, I.; Gemoll, T.; Rody, A.; Thorns, C.; Gertz, E.M.; et al. Single Cell Genetic Profiling of Tumors of Breast Cancer Patients Aged 50 Years and Older Reveals Enormous Intratumor Heterogeneity Independent of Individual Prognosis. Cancers 2021, 13, 3366. https://doi.org/10.3390/cancers13133366
Liegmann A-S, Heselmeyer-Haddad K, Lischka A, Hirsch D, Chen W-D, Torres I, Gemoll T, Rody A, Thorns C, Gertz EM, et al. Single Cell Genetic Profiling of Tumors of Breast Cancer Patients Aged 50 Years and Older Reveals Enormous Intratumor Heterogeneity Independent of Individual Prognosis. Cancers. 2021; 13(13):3366. https://doi.org/10.3390/cancers13133366
Chicago/Turabian StyleLiegmann, Anna-Sophie, Kerstin Heselmeyer-Haddad, Annette Lischka, Daniela Hirsch, Wei-Dong Chen, Irianna Torres, Timo Gemoll, Achim Rody, Christoph Thorns, Edward Michael Gertz, and et al. 2021. "Single Cell Genetic Profiling of Tumors of Breast Cancer Patients Aged 50 Years and Older Reveals Enormous Intratumor Heterogeneity Independent of Individual Prognosis" Cancers 13, no. 13: 3366. https://doi.org/10.3390/cancers13133366