
CD33 Expression and Gentuzumab Ozogamicin in Acute Myeloid Leukemia: Two Sides of the Same Coin

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. CD33 Expression in Normal Myelopoiesis
2. CD33 Expression in Acute Myeloid Leukemia
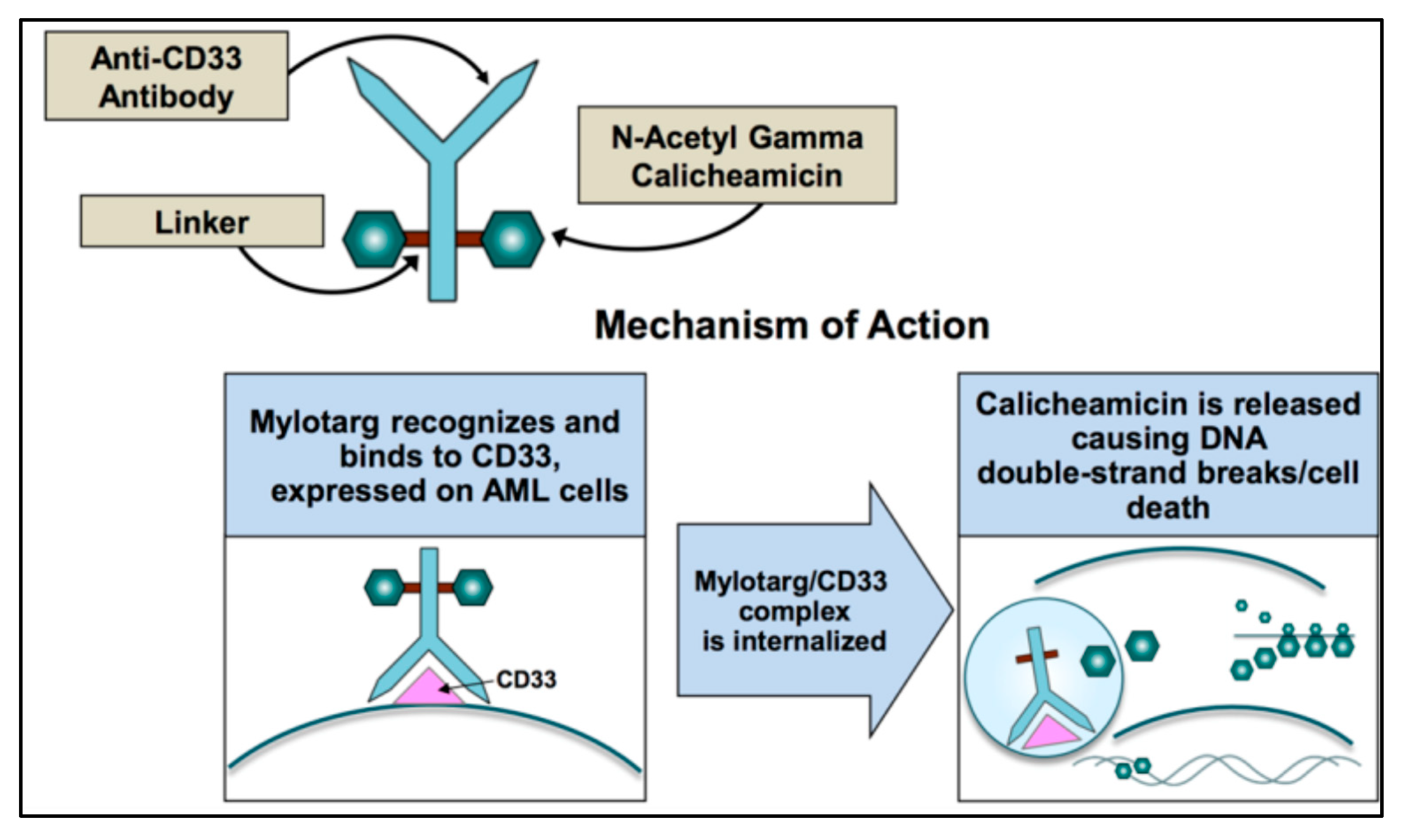
3. Gemtuzumab Ozogamicin (GO) and Mechanism of Action
4. GO and Clinical Studies

{kind=link}
| Study | Treatment (Induction) | Patients | Median Age (Years) | ORR (CR + CRi) | OS | PFS/EFS | Early Mortality | Relapse | Comment | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| AAML0531 | ADE (10 + 3 + 5) +/− GO 3 mg/m2 D + 6 | 1022 | 9.7 (0–30) | 88% v 85%; | 3 years: (69.4% v 65.4%; p = 0.39) | EFS (3 yrs): 53.1% v 46.9%; p = 0.04 | 6.6% v 4.1%; p = 0.09 | 3 yrs: 32.8% v 41.3%; p = 0.006 | In pediatric patients, low doses of GO did not increase OS. EFS was improved with fewer relapses but slightly increased toxicity. | [59] |
| SWOG S0106 | DA (dauno 45 mg/m2) + GO 6 mg/m2 D4 vs. DA (dauno 60 mg/m2) | 595 | 47(18–60) | (69% v 70% p = 0.59 +76% v 74% p = 0.36 | 5 years: 46% v 50% p = 0.85 | RFS (5 yrs) 43% v 42%; p = 0.4 | 5% v 1% p = 0.0062 | 5 yrs: 43% v 42%; p = 0.4 | GO failed to show improvement in CR rate, DFS, or OS. Toxicity was significantly higher. | [42] |
| ALFA-0701 | DA + GO 3 mg/m2 D 1,4,7 vs. DA | 271 | 62.2 | CR: 70.4% v 69.9%; CRp: 11.1% v 3.7% | Median 27.5 v 21.8 moths p = 0.16 | Median EFS 17.3 v 9.5 months p = 0.0002. No advantage in EFS with GO for poor cytogenetic risk | 6% v 4% | Median RFS 28.0 v 11.4 months | Fractionation of doses of the GO allows safe delivery of a much higher cumulative dose and improves outcomes. | [49,50] |
| MRC AML15 | DA or FLAG-Ida +/− GO 3 mg/m2 D + 1 | 1113 | 49 (0–71) | CR: 82% v 83% p = 0.8; CRi: 3% v 4% p = 0.4 | 5 years: 43% v 41% p = 0.3 OS improved with GO for good risk cytogenetics | RFS (5 yrs) 39% v 35%; p = 0.09 | 11% v 10% | 5-YRS 46% v 50% p = 0.12 | A single low dose of GO associated with different induction schemes in young patients produced similar outcomes. However, a survival benefit for patients with favorable cytogenetics was evident. | [43] |
| NCRI AML16 | DA (3 + 10) or DClofarabine +/−GO 3 mg/m2 D + 1 | 1115 | 67 (51–84) | CR: 62% v 58% p = 0.14; CRi: 9% v 10% p = 0.3 | 3 years: (25% v 20%; p = 0.05) | RFS (3 yrs) 21% v 16%; p = 0.04 | 9% v 8% | 3-YRS 68% v 76% p = 0.007 | Single low dose of GO in older pts. significantly reduced relapse risk, and improved OS with acceptable toxicity | [44] |
| NCRI AML17 | ADE or DA + GO 3 mg/m2 v 6 mg/m2 | 788 | 50 (0–81) | CR: 82% v 76% p = 0.003; CRi: 7% v 10% p = 0.17 | 4 years: (50% v 47%; p = 0.3) | RFS (4 yrs) 44% v 38%; p = 0.3 | 3% v 7%; p = 0.02 | 4-YRS 46% v 54% p = 0.15 | Single low dose of GO had similar disease-free and overall survival, but less toxicity with respect to intermediate dose. | [52] |
| GOELAMS AML 2006 IR | DA +/− GO 6 mg/m2 | 238 | 50 (18–60) | 91.6% v 86.5% (p = NS) | 3 years: 53% v 46% | EFS (3 yrs) 51% v 33% | 10% v 4.5% (p = NS) | / | In patients with intermediate cytogenetics AML, GO failed to improve OS | [45,46] |
| EORTC-GIMEMA AML-17 | MICE +/− GO 6 mg/m2 D 1, 15 | 472 | 67 (60–75) | CR: 39% v 41%; CRp: 9% v 8% | 2.5 years:16% v 21.7% p = 0.07 | EFS (1 yr) 18% | 17% v 12% | / | Combining two upfront doses of GO 6 mg/m2 with sequential chemotherapy does not benefit older patients with AML, is too toxic for those >70 years. | [56] |
| EORTC-GIMEMA AML-19 | GO (6 mg/m2 on D1 and 3 mg/m2 on D8) vs. BSC | 237 | 77 (62–88) | CR: 8.1% CRi: 16.2% | 1 year: 24.3% v 9.7% | Median DFS was 5.3 months | 7% | / | Older patients treated in first line with GO showed significantly improved OS in all subgroups, with comparable toxicity than BSC. | [57] |
5. GO and Current Indications
6. In Vitro Relationship between CD33 Expression and GO Efficacy
7. In Vivo Relationship between CD33 Expression and GO Efficacy
8. Prognostic Impact of Cytogenetic Alterations and Molecular Profile on GO Efficacy
9. Relationship between CD33 Single Nucleotide Polymorphisms and GO Efficacy
10. GO Resistance
11. GO as Maintenance Therapy in AML
12. CD33 Bispecific Antibodies and CAR-T CD33
13. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Freeman, S.; Kelm, S.; Barber, E.; Crocker, P. Characterization of CD33 as a new member of the sialoadhesin family of cellular interaction molecules. Blood 1995, 85, 2005–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocker, P.R. Siglecs: Sialic-acid-binding immunoglobulin-like lectins in cell–cell interactions and signalling. Curr. Opin. Struct. Biol. 2002, 12, 609–615. [Google Scholar] [CrossRef]
- Walter, R.B.; Gooley, T.A.; Van Der Velden, V.H.J.; Loken, M.R.; Van Dongen, J.J.M.; Flowers, D.A.; Bernstein, I.D.; Appelbaum, F.R. CD33 expression and P-glycoprotein–mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood 2007, 109, 4168–4170. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Macauley, M.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Balaian, L.; Zhong, R.-K.; Ball, E.D. The inhibitory effect of anti-CD33 monoclonal antibodies on AML cell growth correlates with Syk and/or ZAP-70 expression. Exp. Hematol. 2003, 31, 363–371. [Google Scholar] [CrossRef]
- Balaian, L.; Ball, E.D. Direct effect of bispecific anti-CD33 × anti-CD64 antibody on proliferation and signaling in myeloid cells. Leuk. Res. 2001, 25, 1115–1125. [Google Scholar] [CrossRef]
- Hernández-Caselles, T.; Martínez-Esparza, M.; Pérez-Oliva, A.B.; Quintanilla-Cecconi, A.M.; García-Alonso, A.; Alvarez-López, D.M.R.; García-Peñarrubia, P. A study of CD33 (SIGLEC-3) antigen expression and function on activated human T and NK cells: Two isoforms of CD33 are generated by alternative splicing. J. Leukoc. Biol. 2006, 79, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.; Torok-Storb, B.; Bernstein, I. Myeloid-associated differentiation antigens on stem cells and their progeny identified by monoclonal antibodies. Blood 1983, 62, 124–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brendel, C.; Neubauer, A. Characteristics and analysis of normal and leukemic stem cells: Current concepts and future directions. Leukemia 2000, 14, 1711–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmi, C.; Martelli, M.P.; Diverio, D.; Fenu, S.; Vegna, M.L.; Cantù-Rajnoldi, A.; Biondi, A.; Cocito, M.G.; Del Vecchio, L.; Tabilio, A.; et al. Immunophenotype of adult and childhood acute promyelocytic leukaemia: Correlation with morphology, type of PML gene breakpoint and clinical outcome. A cooperative Italian study on 196 cases. Br. J. Haematol. 1998, 102, 1035–1041. [Google Scholar] [CrossRef]
- Jilani, I.; Estey, E.; Huh, Y.; Joe, Y.; Manshouri, T.; Yared, M.; Giles, F.; Kantarjian, H.; Cortes, J.; Thomas, D.; et al. Differences in CD33 Intensity Between Various Myeloid Neoplasms. Am. J. Clin. Pathol. 2002, 118, 560–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handgretinger, R.; Schäfer, H.-J.; Baur, F.; Frank, D.; Ottenlinger, C.; Bühring, H.-J.; Niethammer, D. Expression of an early myelopoietic antigen (CD33) on a subset of human umbilical cord blood-derived natural killer cells. Immunol. Lett. 1993, 37, 223–228. [Google Scholar] [CrossRef]
- Raponi, S.; De Propris, M.S.; Intoppa, S.; Milani, M.L.; Vitale, A.; Elia, L.; Perbellini, O.; Pizzolo, G.; Foa, R.; Guarini, A. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: Analysis of 552 cases. Leuk. Lymphoma 2011, 52, 1098–1107. [Google Scholar] [CrossRef]
- Walter, R.B.; Appelbaum, F.R.; Estey, E.H.; Bernstein, I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012, 119, 6198–6208. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.A.; Alonzo, T.A.; Loken, M.; Gerbing, R.B.; Ho, P.A.; Bernstein, I.D.; Raimondi, S.C.; Hirsch, B.; Franklin, J.; Walter, R.B.; et al. Correlation of CD33 expression level with disease characteristics and response to gemtuzumab ozogamicin containing chemotherapy in childhood AML. Blood 2012, 119, 3705–3711. [Google Scholar] [CrossRef] [Green Version]
- Olombel, G.; Guerin, E.; Guy, J.; Perrot, J.-Y.; Dumézy, F.; De Labarthe, A.; Bastie, J.-N.; Legrand, O.; Raffoux, E.; Plesa, A.; et al. The level of blast CD33 expression positively impacts the effect of gemtuzumab ozogamicin in patients with acute myeloid leukemia. Blood 2016, 127, 2157–2160. [Google Scholar] [CrossRef]
- Khan, N.; Hills, R.; Virgo, P.; Couzens, S.; Clark, N.; Gilkes, A.; Richardson, P.; Knapper, S.; Grimwade, D.; Russell, N.H.; et al. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leukemia 2017, 31, 1059–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinndorf, P.A.; Buckley, J.D.; Nesbit, M.E.; Lampkin, B.C.; Piomelli, S.; Feig, S.A.; Kersey, J.H.; Hammond, G.D.; Bernstein, I.D. Expression of myeloid differentiation antigens in acute nonlymphocytic leukemia: Increased concentration of CD33 antigen predicts poor outcome—A report from the childrens cancer study group. Med. Pediatr. Oncol. 1992, 20, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, B.; Gil, D.; Bowen, D.T.; Crocker, P.R. Analysis of the CD33-related siglec family reveals that Siglec-9 is an endocytic receptor expressed on subsets of acute myeloid leukemia cells and absent from normal hematopoietic progenitors. Leuk. Res. 2020, 31, 211–220. [Google Scholar] [CrossRef]
- Friedrich, M.; Henn, A.; Raum, T.; Bajtus, M.; Matthes, K.; Hendrich, L.; Wahl, J.; Hoffmann, P.; Kischel, R.; Kvesic, M.; et al. Preclinical Characterization of AMG 330, a CD3/CD33-Bispecific T-Cell–Engaging Antibody with Potential for Treatment of Acute Myelogenous Leukemia. Mol. Cancer Ther. 2014, 13, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab Ozogamicin, A Potent and Selective Anti-CD33 Antibody−Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Bioconjugate. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Lancet, J.E.; Wood, B.L.; Grove, L.E.; Sandalic, L.; Sievers, E.; Jurcic, J.G. Randomized, phase IIb study of low-dose cytarabine and lintuzumab versus low-dose cytarabine and placebo in older adults with untreated acute myeloid leukemia. Haematologica 2012, 98, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Van Der Velden, V.H.J.; Te Marvelde, J.G.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; van Dongen, J.J. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204. [Google Scholar] [CrossRef]
- Godwin, C.D.; McDonald, G.B.; Walter, R.B. Sinusoidal obstruction syndrome following CD33-targeted therapy in acute myeloid leukemia. Blood 2017, 129, 2330–2332. [Google Scholar] [CrossRef] [Green Version]
- Lamba, J.K.; Pounds, S.; Cao, X.; Downing, J.R.; Campana, D.; Ribeiro, R.C.; Pui, C.H.; Rubnitz, J.E. Coding polymorphisms in CD33 and response to gemtuzumab ozogamicin in pediatric patients with AML: A pilot study. Leukemia 2009, 23, 402–404. [Google Scholar] [CrossRef] [Green Version]
- Hamann, P.R.; Hinman, L.M.; Beyer, C.F.; Lindh, D.; Upeslacis, J.; Flowers, D.A.; Bernstein, I. An anti-CD33 antibody—Calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconjug. Chem. 2002, 13, 40–46. [Google Scholar] [CrossRef]
- Boyer, T.; Gonzales, F.; Barthélémy, A.; Marceau-Renaut, A.; Peyrouze, P.; Guihard, S.; Lepelley, P.; Plesa, A.; Nibourel, O.; Delattre, C.; et al. Clinical Significance of ABCB1 in Acute Myeloid Leukemia: A Comprehensive Study. Cancers 2019, 11, 1323. [Google Scholar] [CrossRef] [Green Version]
- Larson, R.A.; Sievers, E.; Stadtmauer, E.A.; Löwenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef]
- Sievers, E.; Larson, R.; Stadtmauer, E.A.; Estey, E.; Löwenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L.; et al. Efficacy and Safety of Gemtuzumab Ozogamicin in Patients With CD33-Positive Acute Myeloid Leukemia in First Relapse. J. Clin. Oncol. 2001, 19, 3244–3254. [Google Scholar] [CrossRef] [PubMed]
- Linenberger, M.L.; Hong, T.; Flowers, D.; Sievers, E.L.; Gooley, T.A.; Bennett, J.M.; Berger, M.S.; Leopold, L.H.; Appelbaum, F.R.; Bernstein, I.D. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 2001, 98, 988–994. [Google Scholar] [CrossRef] [Green Version]
- Yaghoubi, S.; Karimi, M.H.; Lotfinia, M.; Gharibi, T.; Mahi-Birjand, M.; Kavi, E.; Hosseini, F.; Sepehr, K.S.; Khatami, M.; Bagheri, N.; et al. Potential drugs used in the antibody–drug conjugate (ADC) architecture for cancer therapy. J. Cell. Physiol. 2020, 235, 31–64. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.R.; Ju, J.; Shen, B.; Beerman, T.A. Designer enediynes generate DNA breaks, interstrand cross-links, or both, with concomitant changes in the regulation of DNA damage responses. Proc. Natl. Acad. Sci. USA 2007, 104, 17632–17637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, N.; Lyne, L. Sensitivity of fibroblasts derived from ataxia-telangiectasia patients to calicheamicin γ1I. Mutat. Res. Lett. 1990, 245, 171–175. [Google Scholar] [CrossRef]
- Prokop, A.; Wrasidlo, W.; Lode, H.; Lang, F.; Henze, H.; Dörken, B.; Wieder, T.; Daniel, P.T. Induction of apoptosis by enediyne antibiotic calicheamicin θII proceeds through a caspase-mediated mitochondrial amplification loop in an entirely Bax-dependent manner. Oncogene 2003, 22, 9107–9120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, M.; Aldoss, I.; Marcucci, G.; Pullarkat, V. Hypomethylating agents in combination with venetoclax for acute myeloid leukemia: Update on clinical trial data and practical considerations for use. Am. J. Hematol. 2018, 94, 358–362. [Google Scholar] [CrossRef]
- Godwin, C.D.; Bates, O.M.; Jean, S.R.; Laszlo, G.S.; Garling, E.E.; Beddoe, M.E.; Cardone, M.H.; Walter, R.B. Anti-apoptotic BCL-2 family proteins confer resistance to calicheamicin-based antibody-drug conjugate therapy of acute leukemia. Leuk. Lymphoma 2020, 61, 2990–2994. [Google Scholar] [CrossRef]
- Goemans, B.F.; Zwaan, C.M.; Vijverberg, S.J.H.; Loonen, A.H.; Creutzig, U.; Hählen, K.; Reinhardt, D.; Gibson, B.E.S.; Cloos, J.; Kaspers, G.J.L. Large interindividual differences in cellular sensitivity to calicheamicin may influence gemtuzumab ozogamicin response in acute myeloid leukemia. Leukemia 2008, 22, 2284–2285. [Google Scholar] [CrossRef] [Green Version]
- Sievers, E.L.; Appelbaum, F.R.; Spielberger, R.T.; Forman, S.J.; Flowers, D.; Smith, F.O.; Shannon-Dorcy, K.; Berger, M.S.; Bernstein, I.D. Selective Ablation of Acute Myeloid Leukemia Using Antibody-Targeted Chemotherapy: A Phase I Study of an Anti-CD33 Calicheamicin Immunoconjugate. Blood 1999, 93, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar] [PubMed]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [Green Version]
- Burnett, A.K.; Hills, R.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of Patients With Acute Myeloblastic Leukemia Who Benefit From the Addition of Gemtuzumab Ozogamicin: Results of the MRC AML15 Trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef]
- Burnett, A.K.; Russell, N.H.; Hills, R.; Kell, J.; Freeman, S.; Kjeldsen, L.; Hunter, A.E.; Yin, J.; Craddock, C.F.; Dufva, I.H.; et al. Addition of Gemtuzumab Ozogamicin to Induction Chemotherapy Improves Survival in Older Patients With Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 3924–3931. [Google Scholar] [CrossRef]
- Delaunay, J.; Recher, C.; Pigneux, A.; Witz, F.; Vey, N.; Blanchet, O.; Lefebvre, P.; Luquet, I.; Guillerme, I.; Volteau, C.; et al. Addition of Gemtuzumab Ozogamycin to Chemotherapy Improves Event-Free Survival but Not Overall Survival of AML Patients with Intermediate Cytogenetics Not Eligible for Allogeneic Transplantation. Results of the GOELAMS AML 2006 IR Study. Blood 2011, 118, 79. [Google Scholar] [CrossRef]
- Bouvier, A.; Hamel, J.; Delaunay, J.; Delabesse, E.; Dumas, P.Y.; Ledoux, M.P.; Peterlin, P.; Luquet, I.; Guepin, G.R.; Bulabois, C.E.; et al. Molecular classification and prognosis in younger adults with acute myeloid leukemia patients and intermediate-risk cytogenetics treated or not by gemtuzumab ozogamycin: Final results of the GOELAMS/FILO AML 2006-IR trial. Eur. J. Haematol. 2021. [Google Scholar] [CrossRef]
- Taksin, A.-L.; Legrand, O.; Raffoux, E.; De Revel, T.; Thomas, X.; Contentin, N.; Bouabdallah, R.; Pautas, C.; Turlure, P.; Reman, O.; et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: A prospective study of the alfa group. Leukemia 2006, 21, 66–71. [Google Scholar] [CrossRef]
- Farhat, H.; Reman, O.; Raffoux, E.; Berthon, C.; Pautas, C.; Kammoun, L.; Chantepie, S.; Gardin, C.; Rousselot, P.; Chevret, S.; et al. Fractionated doses of gemtuzumab ozogamicin with escalated doses of daunorubicin and cytarabine as first acute myeloid leukemia salvage in patients aged 50–70-year old: A phase 1/2 study of the acute leukemia French association. Am. J. Hematol. 2012, 87, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Lambert, J.; Pautas, C.; Terré, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2019, 104, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Burnett, A.; Cavenagh, J.; Russell, N.; Hills, R.; Kell, J.; Jones, G.; Nielsen, O.J.; Khwaja, A.; Thomas, I.; Clark, R.; et al. Defining the dose of gemtuzumab ozogamicin in combination with induction chemotherapy in acute myeloid leukemia: A comparison of 3 mg/m2 with 6 mg/m2 in the NCRI AML17 Trial. Haematologica 2016, 101, 724–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hills, R.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Schlenk, R.F.; Paschka, P.; Krzykalla, J.; Weber, D.; Kapp-Schwoerer, S.; Gaidzik, V.I.; Leis, C.; Fiedler, W.; Kindler, T.; Schroeder, T.; et al. Gemtuzumab Ozogamicin in NPM1-Mutated Acute Myeloid Leukemia: Early Results From the Prospective Randomized AMLSG 09-09 Phase III Study. J. Clin. Oncol. 2020, 38, 623–632. [Google Scholar] [CrossRef]
- Fournier, E.; Duployez, N.; Ducourneau, B.; Raffoux, E.; Turlure, P.; Caillot, D.; Thomas, X.; Marceau-Renaut, A.; Chantepie, S.P.; Malfuson, J.-V.; et al. Mutational profile and benefit of gemtuzumab ozogamicin in acute myeloid leukemia. Blood 2020, 135, 542–546. [Google Scholar] [CrossRef]
- Amadori, S.; Suciu, S.; Stasi, R.; Salih, H.R.; Selleslag, D.; Muus, P.; De Fabritiis, P.; Venditti, A.; Ho, A.D.; Lübbert, M.; et al. Sequential Combination of Gemtuzumab Ozogamicin and Standard Chemotherapy in Older Patients with Newly Diagnosed Acute Myeloid Leukemia: Results of a Randomized Phase III Trial by the EORTC and GIMEMA Consortium (AML-17). J. Clin. Oncol. 2013, 31, 4424–4430. [Google Scholar] [CrossRef]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab Ozogamicin versus Best Supportive Care in Older Patients with Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III EORTC-GIMEMA AML-19 Trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Pautas, C.; Raffoux, E.; Lambert, J.; Legrand, O.; Chantepie, S.; Gastaud, L.; Marolleau, J.-P.; Thomas, X.; Turlure, P.; Benner, R.J.; et al. Outcomes following hematopoietic stem cell transplantation in patients treated with standard chemotherapy with or without gemtuzumab ozogamicin for acute myeloid leukemia. Bone Marrow Transplant. 2021, 56, 1474–1477. [Google Scholar] [CrossRef]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab Ozogamicin in Children and Adolescents with De Novo Acute Myeloid Leukemia Improves Event-Free Survival by Reducing Relapse Risk: Results from the Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2014, 32, 3021–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA. MYLOTARGTM (Gemtuzumab Ozogamicin) for Injection. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761060lbl.pdf (accessed on 17 October 2020).
- Norsworthy, K.J.; Ko, C.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Dunmore, H.; Karres, D.; Hay, J.L.; Salmonsson, T.; Gisselbrecht, C.; Sarac, S.B.; Bjerrum, O.W.; Hovgaard, D.; Barbachano, Y.; et al. The EMA Review of Mylotarg (Gemtuzumab Ozogamicin) for the Treatment of Acute Myeloid Leukemia. Oncologist 2019, 24, e171–e179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EMA. EPAR—Summary for Mylotarg—Epar Product Information Public. Available online: https://www.ema.europa.eu/en/documents/product-information/mylotarg-epar-product-information_en.pdf (accessed on 17 October 2020).
- Walter, R.B.; Raden, B.W.; Kamikura, D.M.; Cooper, J.A.; Bernstein, I.D. Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab ozogamicin–induced cytotoxicity. Blood 2005, 105, 1295–1302. [Google Scholar] [CrossRef] [Green Version]
- Jawad, M.; Seedhouse, C.; Mony, U.; Grundy, M.; Russell, N.H.; Pallis, M. Analysis of factors that affect in vitro chemosensitivity of leukaemic stem and progenitor cells to gemtuzumab ozogamicin (Mylotarg) in acute myeloid leukaemia. Leukemia 2009, 24, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Chevallier, P.; Delaunay, J.; Turlure, P.; Pigneux, A.; Hunault, M.; Garand, R.; Guillaume, T.; Avet-Loiseau, H.; Dmytruk, N.; Girault, S.; et al. Long-Term Disease-Free Survival after Gemtuzumab, Intermediate-Dose Cytarabine, and Mitoxantrone in Patients With CD33+Primary Resistant or Relapsed Acute Myeloid Leukemia. J. Clin. Oncol. 2008, 26, 5192–5197. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.A.; Loken, M.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Aplenc, R.; Bernstein, I.D.; Gamis, A.S.; Alonzo, T.A.; Meshinchi, S. CD33 Expression and Its Association with Gemtuzumab Ozogamicin Response: Results From the Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2016, 34, 747–755. [Google Scholar] [CrossRef]
- Wiemels, J.L.; Xiao, Z.; Buffler, P.A.; Maia, A.-T.; Ma, X.; Dicks, B.M.; Smith, M.T.; Zhang, L.; Feusner, J.; Wiencke, J.; et al. In utero origin of t(8;21) AML1-ETO translocations in childhood acute myeloid leukemia. Blood 2002, 99, 3801–3805. [Google Scholar] [CrossRef]
- Jourdan, E.; Boissel, N.; Chevret, S.; Delabesse, E.; Renneville, A.; Cornillet, P.; Blanchet, O.; Cayuela, J.-M.; Recher, C.; Raffoux, E.; et al. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood 2013, 121, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Appelbaum, F.R.; Bernstein, I.D. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood 2017, 130, 2373–2376. [Google Scholar] [CrossRef] [Green Version]
- Munoz, L.E.; Nomdedeu, J.; Villamor, N.; De La Guardia, R.M.M.; Colomer, D.; Ribera, J.-M.; Torres, J.P.; Berlanga, J.J.; Fernandez, C.; Llorente, A.; et al. Acute myeloid leukemia with MLL rearrangements: Clinicobiological features, prognostic impact and value of flow cytometry in the detection of residual leukemic cells. Leukemia 2003, 17, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, H.; Yamamoto, G.; Hosoi, M.; Takahashi, T.; Hangaishi, A.; Kurokawa, M. Complete molecular remission in refractory acute myeloid leukemia with MLL/AF9 treated with gemtuzumab ozogamicin. Leuk. Res. 2010, 34, e152–e153. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, A.; Shinjo, K.; Naito, K.; Matsui, H.; Sahara, N.; Shigeno, K.; Horii, T.; Shirai, N.; Maekawa, M.; Ohnishi, K.; et al. Efficacy of gemtuzumab ozogamicin on ATRA- and arsenic-resistant acute promyelocytic leukemia (APL) cells. Leukemia 2005, 19, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, A.; Shinjo, K.; Naito, K.; Matsui, H.; Sahara, N.; Shigeno, K.; Suzumura, T.; Horii, T.; Shirai, N.; Maekawa, M.; et al. Two Patients with All-trans Retinoic Acid-Resistant Acute Promyelocytic Leukemia Treated Successfully with Gemtuzumab Ozogamicin as a Single Agent. Int. J. Hematol. 2005, 82, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Estey, E.; Jones, D.; Faderl, S.; O’Brien, S.; Fiorentino, J.; Pierce, S.; Blamble, D.; Estrov, Z.; Wierda, W.; et al. Effective Treatment of Acute Promyelocytic Leukemia with All-Trans-Retinoic Acid, Arsenic Trioxide, and Gemtuzumab Ozogamicin. J. Clin. Oncol. 2009, 27, 504–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancet, J.E.; Moseley, A.B.; Coutre, S.E.; DeAngelo, D.; Othus, M.; Tallman, M.S.; Litzow, M.R.; Komrokji, R.S.; Erba, H.P.; Appelbaum, F.R. A phase 2 study of ATRA, arsenic trioxide, and gemtuzumab ozogamicin in patients with high-risk APL (SWOG 0535). Blood Adv. 2020, 4, 1683–1689. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renneville, A.; Ben Abdelali, R.; Chevret, S.; Nibourel, O.; Cheok, M.; Pautas, C.; Duléry, R.; Boyer, T.; Cayuela, J.-M.; Hayette, S.; et al. Clinical impact of gene mutations and lesions detected by SNP-array karyotyping in acute myeloid leukemia patients in the context of gemtuzumab ozogamicin treatment: Results of the ALFA-0701 trial. Oncotarget 2014, 5, 916–932. [Google Scholar] [CrossRef] [Green Version]
- Tarlock, K.; Alonzo, T.A.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Sung, L.; Pollard, J.A.; Aplenc, R.; Loken, M.R.; Gamis, A.S.; et al. Gemtuzumab Ozogamicin Reduces Relapse Risk in FLT3/ITD Acute Myeloid Leukemia: A Report from the Children’s Oncology Group. Clin. Cancer Res. 2016, 22, 1951–1957. [Google Scholar] [CrossRef] [Green Version]
- Mortland, L.; Alonzo, T.A.; Walter, R.; Gerbing, R.B.; Mitra, A.; Pollard, J.A.; Loken, M.R.; Hirsch, B.; Raimondi, S.; Franklin, J.; et al. Clinical Significance of CD33 Nonsynonymous Single-Nucleotide Polymorphisms in Pediatric Patients with Acute Myeloid Leukemia Treated with Gemtuzumab-Ozogamicin–Containing Chemotherapy. Clin. Cancer Res. 2013, 19, 1620–1627. [Google Scholar] [CrossRef] [Green Version]
- Lamba, J.K.; Chauhan, L.; Shin, M.; Loken, M.R.; Pollard, J.A.; Wang, Y.-C.; Ries, R.E.; Aplenc, R.; Hirsch, B.A.; Raimondi, S.C.; et al. CD33 Splicing Polymorphism Determines Gemtuzumab Ozogamicin Response in De Novo Acute Myeloid Leukemia: Report From Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2017, 35, 2674–2682. [Google Scholar] [CrossRef]
- Gale, R.E.; Popa, T.; Wright, M.; Khan, N.; Freeman, S.D.; Burnett, A.K.; Russell, N.H.; Hills, R.; Linch, D.C. No evidence that CD33 splicing SNP impacts the response to GO in younger adults with AML treated on UK MRC/NCRI trials. Blood 2018, 131, 468–471. [Google Scholar] [CrossRef]
- Chauhan, L.; Shin, M.; Wang, Y.-C.; Loken, M.; Pollard, J.; Aplenc, R.; Hirsch, B.A.; Raimondi, S.; Ries, R.E.; Bernstein, I.D.; et al. CD33_PGx6_Score Predicts Gemtuzumab Ozogamicin Response in Childhood Acute Myeloid Leukemia: A Report From the Children’s Oncology Group. JCO Precis. Oncol. 2019, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kartner, N.; Evernden-Porelle, D.; Bradley, G.; Ling, V. Detection of P-glycoprotein in multidrug-resistant cell lines by monoclonal antibodies. Nat. Cell Biol. 1985, 316, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Pastan, I.; Ambudkar, S.V. P-glycoprotein and multidrug resistance. Curr. Opin. Genet. Dev. 1996, 6, 610–617. [Google Scholar] [CrossRef]
- Baba, M.; Nakanishi, O.; Sato, W.; Saito, A.; Miyama, Y.; Yano, O.; Shimada, S.; Fukazawa, N.; Naito, M.; Tsuruo, T. Relationship between multidrug resistant gene expression and multidrug resistant-reversing effect of MS-209 in various tumor cells. Cancer Chemother. Pharmacol. 1995, 36, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.; Seiter, K.; Kolitz, J.; Stock, W.; Giles, F.; Kalaycio, M.; Zenk, D.; Marcucci, G. A Phase II study of Bcl-2 antisense (oblimersen sodium) combined with gemtuzumab ozogamicin in older patients with acute myeloid leukemia in first relapse. Leuk. Res. 2006, 30, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Raden, B.W.; Cronk, M.R.; Bernstein, I.D.; Appelbaum, F.R.; Banker, D.E.; Bello-Fernandez, C.; Stasakova, J.; Renner, A.; Carballido-Perrig, N.; et al. The peripheral benzodiazepine receptor ligand PK11195 overcomes different resistance mechanisms to sensitize AML cells to gemtuzumab ozogamicin. Blood 2004, 103, 4276–4284. [Google Scholar] [CrossRef]
- Haag, P.; Viktorsson, K.; Lindberg, M.L.; Kanter, L.; Lewensohn, R.; Stenke, L. Deficient activation of Bak and Bax confers resistance to gemtuzumab ozogamicin-induced apoptotic cell death in AML. Exp. Hematol. 2009, 37, 755–766. [Google Scholar] [CrossRef]
- Rosen, D.B.; Harrington, K.H.; Cordeiro, J.A.; Leung, L.Y.; Putta, S.; Lacayo, N.; Laszlo, G.S.; Gudgeon, C.J.; Hogge, D.E.; Hawtin, R.E.; et al. AKT Signaling as a Novel Factor Associated with In Vitro Resistance of Human AML to Gemtuzumab Ozogamicin. PLoS ONE 2013, 8, e53518. [Google Scholar] [CrossRef]
- ten Cate, B.; Samplonius, D.F.; Bijma, T.; de Leij, L.F.M.H.; Helfrich, W.; Bremer, E. The histone deacetylase inhibitor valproic acid potently augments gemtuzumab ozogamicin-induced apoptosis in acute myeloid leukemic cells. Leukemia 2007, 21, 248–252. [Google Scholar] [CrossRef]
- Kell, W.J.; Burnett, A.K.; Chopra, R.Y.; Yin, J.A.L.; Clark, R.E.; Rohatiner, A.; Culligan, D.; Hunter, A.; Prentice, A.G.; Milligan, D.W. A feasibility study of simultaneous administration of gemtuzumab ozogamicin with intensive chemotherapy in induction and consolidation in younger patients with acute myeloid leukemia. Blood 2003, 102, 4277–4283. [Google Scholar] [CrossRef] [PubMed]
- Jedema, I.; Barge, R.M.Y.; Van Der Velden, V.H.J.; Nijmeijer, B.A.; Van Dongen, J.J.M.; Willemze, R.; Falkenburg, J.H.F. Internalization and cell cycle-dependent killing of leukemic cells by Gemtuzumab Ozogamicin: Rationale for efficacy in CD33-negative malignancies with endocytic capacity. Leukemia 2003, 18, 316–325. [Google Scholar] [CrossRef]
- Löwenberg, B.; Beck, J.; Graux, C.; van Putten, W.; Schouten, H.R.; Verdonck, L.; Ferrant, A.; Sonneveld, P.; Jongen-Lavrencic, M.; von Lilienfeld-Toal, M.; et al. Gemtuzumab ozogamicin as postremission treatment in AML at 60 years of age or more: Results of a multicenter phase 3 study. Blood 2010, 115, 2586–2591. [Google Scholar] [CrossRef]
- Nagorsen, D.; Baeuerle, P.A. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp. Cell Res. 2011, 317, 1255–1260. [Google Scholar] [CrossRef]
- Brischwein, K.; Parr, L.; Panz, S.; Volkland, J.; Volkland, J.; Lumsden, J.; Klinger, M.; Locher, M.; Hammond, S.A.; Kiener, P.; et al. Strictly target cell-dependent activation of T cells by bispeci c single-chain antibody constructs of the BiTE class. J. Immunother. 2007, 30, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Bögeholz, J.; Köhnke, T.; Lichtenegger, F.S.; Schneider, S.; Metzeler, K.H.; Fiegl, M. CD33 target validation and sustained depletion of AML blasts in long-term cultures b ythe bispecific T-cell-engaging antibody AMG 330. Blood 2014, 123, 356–365. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Westervelt, P.; Cortes, J.E.; Altman, J.K.; Long, M.; Oehler, V.G.; Gojo, I.; Guenot, J.; Chun, P.; Roboz, G.J. Phase 1 rst-in-human trial of AMV564, a bivalent bispecic (2:2) CD33/CD3 T-cell engager, in patients with relapsed/refractory acute myeloid leukemia (AML). Blood 2019, 134 (Suppl. 1), 8. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, K.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia 2021. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-S.; Wang, Y.; Lv, H.-Y.; Han, Q.-W.; Fan, H.; Guo, B.; Wang, L.-L.; Han, W.-D. Treatment of CD33-directed Chimeric Antigen Receptor-modified T Cells in One Patient with Relapsed and Refractory Acute Myeloid Leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subklewe, M.; Stein, A.; Walter, R.B.; Bhatia, R.; Wei, A.H.; Ritchie, D.; Bücklein, V.; Vachhani, P.; Hindoyan, A.; Agarwal, S.; et al. Preliminary Results from a Phase 1 First-in-Human Study of AMG 673, a Novel Half-Life Extended (HLE) Anti-CD33/CD3 BiTE® (Bispecific T-Cell Engager) in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 2019, 134 (Suppl. 1), 833. [Google Scholar] [CrossRef]

| Predictors of response to GO |
|
| Uncertain predictive role on response to GO |
|
| Unfavorable predictors of response to GO |
|
| Resistance mechanisms to GO |
|
| Most important GO-related adverse events in ALFA-0701 study [49,50] |
|
| CD33 bispecific antibodies in clinical trials |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molica, M.; Perrone, S.; Mazzone, C.; Niscola, P.; Cesini, L.; Abruzzese, E.; de Fabritiis, P. CD33 Expression and Gentuzumab Ozogamicin in Acute Myeloid Leukemia: Two Sides of the Same Coin. Cancers 2021, 13, 3214. https://doi.org/10.3390/cancers13133214
Molica M, Perrone S, Mazzone C, Niscola P, Cesini L, Abruzzese E, de Fabritiis P. CD33 Expression and Gentuzumab Ozogamicin in Acute Myeloid Leukemia: Two Sides of the Same Coin. Cancers. 2021; 13(13):3214. https://doi.org/10.3390/cancers13133214
Chicago/Turabian StyleMolica, Matteo, Salvatore Perrone, Carla Mazzone, Pasquale Niscola, Laura Cesini, Elisabetta Abruzzese, and Paolo de Fabritiis. 2021. "CD33 Expression and Gentuzumab Ozogamicin in Acute Myeloid Leukemia: Two Sides of the Same Coin" Cancers 13, no. 13: 3214. https://doi.org/10.3390/cancers13133214