
Distinct Responsiveness of Tumor-Associated Macrophages to Immunotherapy of Tumors with Different Mechanisms of Major Histocompatibility Complex Class I Downregulation

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
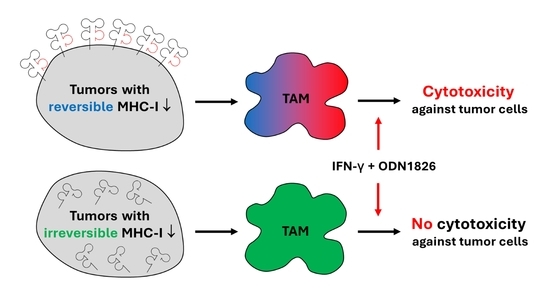
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Tumor Cell Lines
2.3. Plasmid
2.4. Combined IT
2.5. NGS Library Preparation and Sequencing
2.6. RNA-Seq Data Analysis
2.7. Flow Cytometry
2.8. Isolation of TAMs
2.9. In Vitro Stimulations of TAMs
2.10. MTT Cytotoxicity Assay
2.11. NO Measurement
2.12. ELISA
2.13. Arginase Microplate Assay
2.14. Statistical Analysis
3. Results
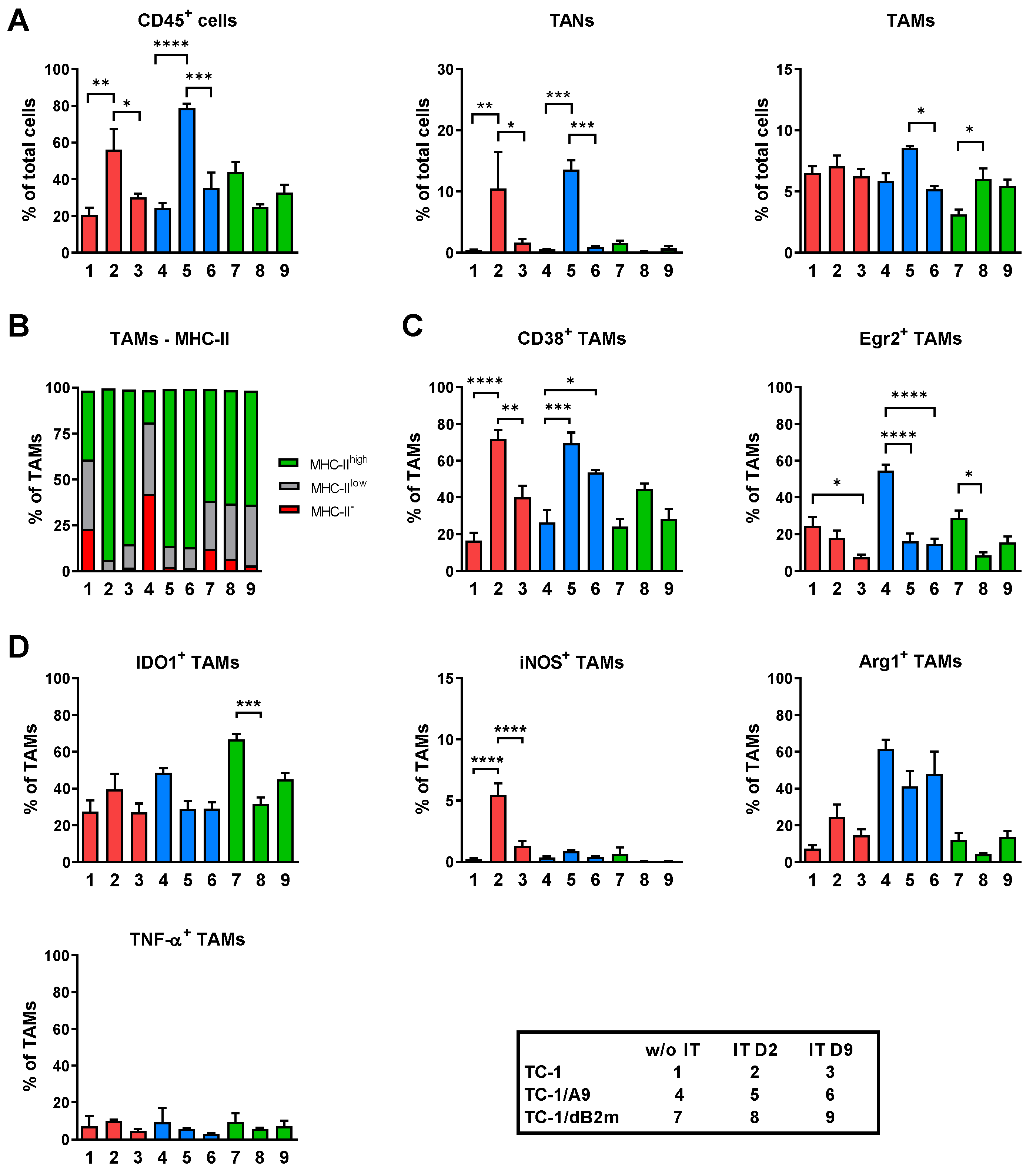
3.1. Microenvironments of TC-1/A9 and TC-1/dB2m Tumors Differ in Immune Reactions
3.2. TAMs from Tumors with Distinct MHC-I Expression Differ in Their Phenotype and Response to IT
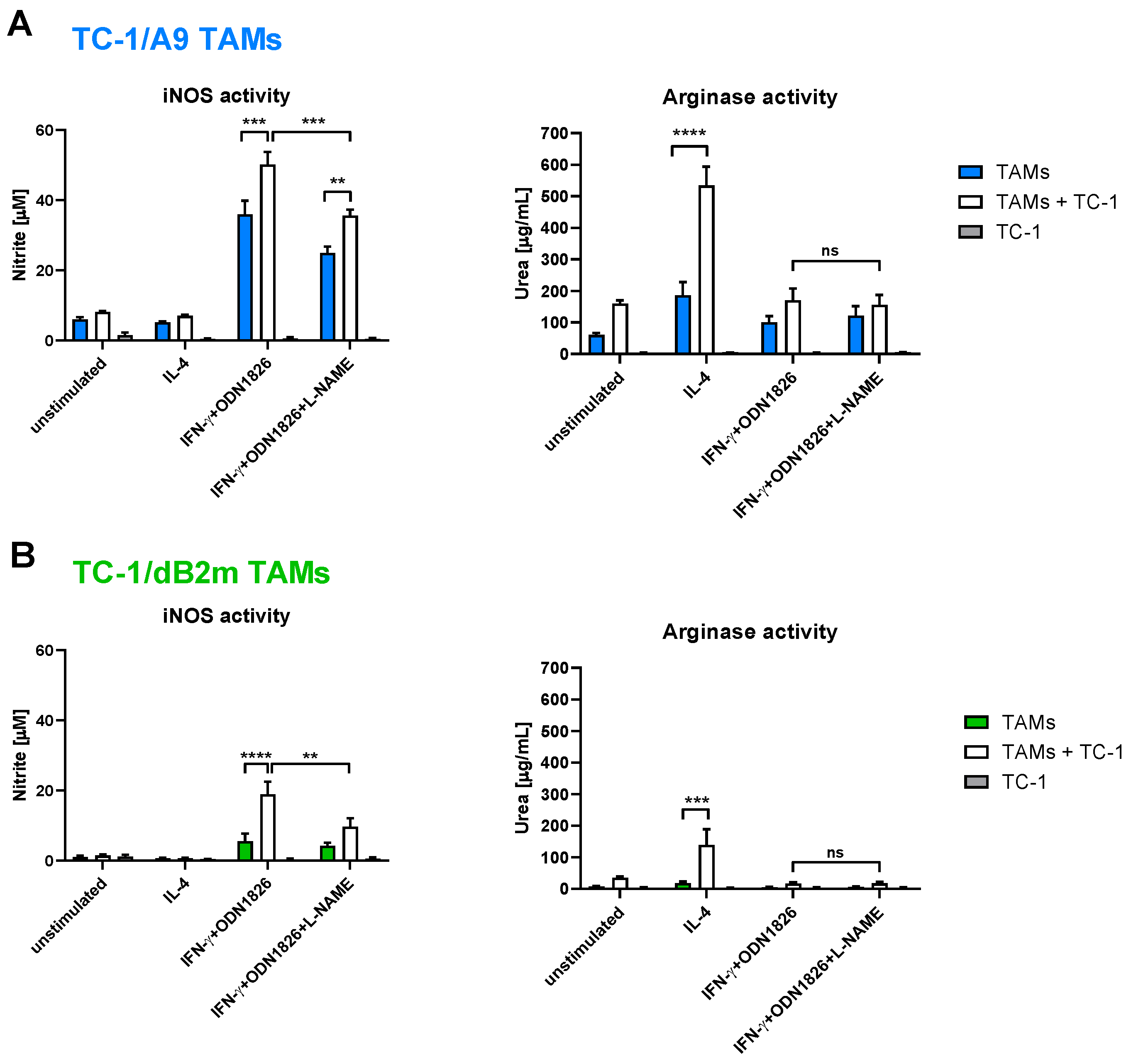
3.3. TAMs from Tumors with Irreversible MHC-I Downregulation Were Not Cytotoxic against Tumor Cells
3.4. In Vitro Inhibition of iNOS Did Not Increase Arginase Activity in TAMs
3.5. TAMs Stimulated with IFN-γ Plus TLR Agonist Produced IL-10 and TNF-α but No IL-12
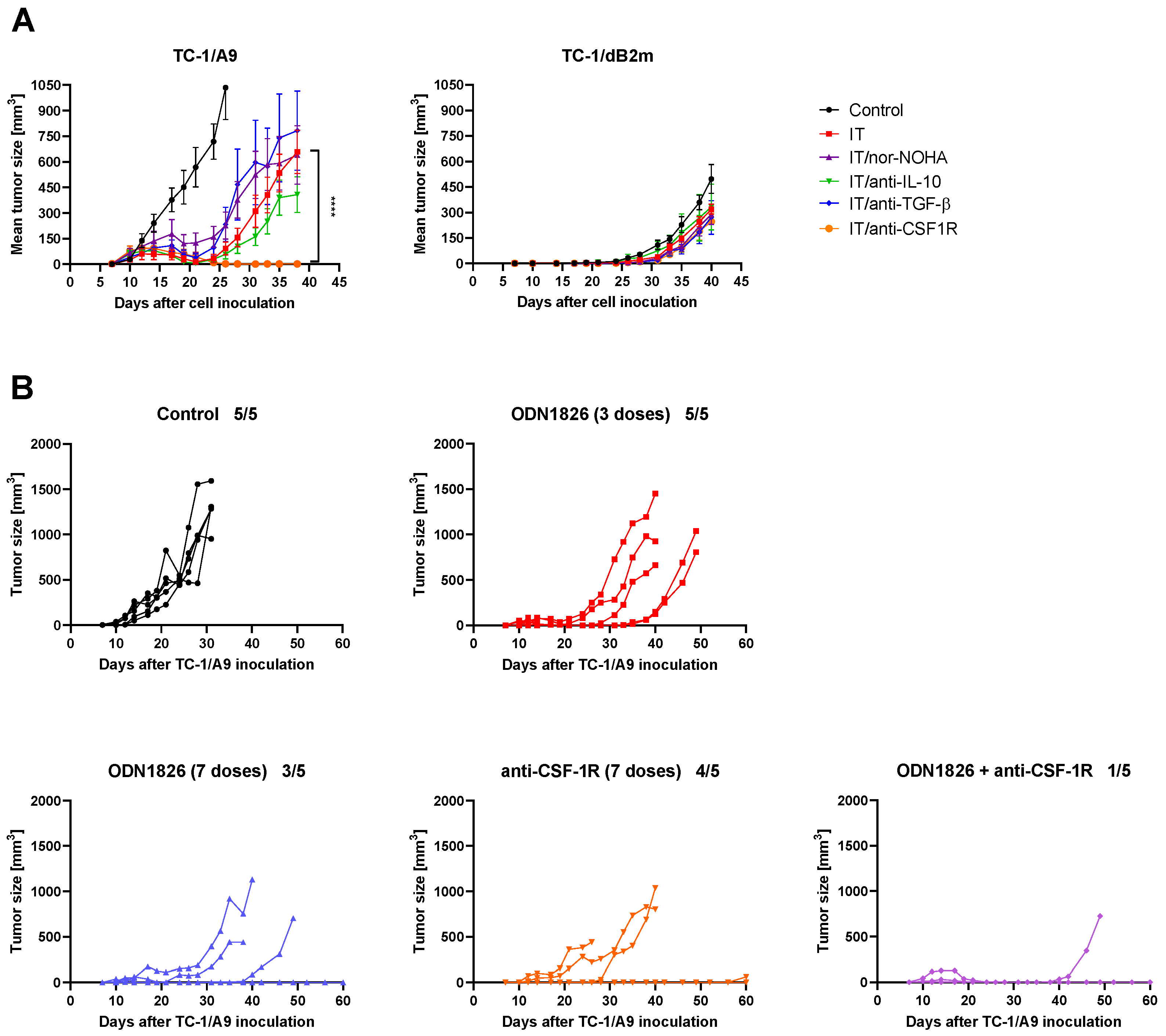
3.6. CSF-1R Blockade Enhanced the Effect of Combined IT But Only in Tumors with Reversible MHC-I Downregulation
3.7. Anti-CSF-1R Barely Affected TAM Repolarization and Slightly Reduced Proportion of CSF-1R-Expressing TAMs in Tumors with Reduced MHC-I Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thor Straten, P.; Garrido, F. Targetless T Cells in Cancer Immunotherapy. J. Immunother. Cancer 2016, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Najafimehr, H.; Hajizadeh, N.; Nazemalhosseini-Mojarad, E.; Pourhoseingholi, M.A.; Abdollahpour-Alitappeh, M.; Ashtari, S.; Zali, M.R. The Role of Human Leukocyte Antigen Class I on Patient Survival in Gastrointestinal Cancers: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 728. [Google Scholar] [CrossRef] [Green Version]
- Schaafsma, E.; Fugle, C.M.; Wang, X.; Cheng, C. Pan-Cancer Association of HLA Gene Expression with Cancer Prognosis and Immunotherapy Efficacy. Br. J. Cancer 2021, 1–11. [Google Scholar] [CrossRef]
- Ryschich, E.; Nötzel, T.; Hinz, U.; Autschbach, F.; Ferguson, J.; Simon, I.; Weitz, J.; Fröhlich, B.; Klar, E.; Büchler, M.W.; et al. Control of T-Cell–Mediated Immune Response by HLA Class I in Human Pancreatic Carcinoma. Clin. Cancer Res. 2005, 11, 498–504. [Google Scholar]
- Goeppert, B.; Frauenschuh, L.; Zucknick, M.; Roessler, S.; Mehrabi, A.; Hafezi, M.; Stenzinger, A.; Warth, A.; Pathil, A.; Renner, M.; et al. Major Histocompatibility Complex Class I Expression Impacts on Patient Survival and Type and Density of Immune Cells in Biliary Tract Cancer. Br. J. Cancer 2015, 113, 1343–1349. [Google Scholar] [CrossRef] [Green Version]
- Perea, F.; Bernal, M.; Sánchez-Palencia, A.; Carretero, J.; Torres, C.; Bayarri, C.; Gómez-Morales, M.; Garrido, F.; Ruiz-Cabello, F. The Absence of HLA Class I Expression in Non-Small Cell Lung Cancer Correlates with the Tumor Tissue Structure and the Pattern of T Cell Infiltration. Int. J. Cancer 2017, 140, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Perea, F.; Bernal, M.; Sánchez-Palencia, A.; Aptsiauri, N.; Ruiz-Cabello, F. The Escape of Cancer from T Cell-Mediated Immune Surveillance: HLA Class I Loss and Tumor Tissue Architecture. Vaccines 2017, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Khong, H.; Dai, Z.; Huang, X.-F.; Wargo, J.A.; Cooper, Z.A.; Vasilakos, J.P.; Hwu, P.; Overwijk, W.W. Effective Innate and Adaptive Antimelanoma Immunity through Localized TLR7/8 Activation. J. Immunol. 2014, 193, 4722–4731. [Google Scholar] [CrossRef] [Green Version]
- Moynihan, K.D.; Irvine, D.J. Roles for Innate Immunity in Combination Immunotherapies. Cancer Res. 2017, 77, 5215–5221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhmilevich, A.L.; Felder, M.; Lever, L.; Slowinski, J.; Rasmussen, K.; Hoefges, A.; van de Voort, T.J.; Loibner, H.; Korman, A.J.; Gillies, S.D.; et al. Effective Combination of Innate and Adaptive Immunotherapeutic Approaches in a Mouse Melanoma Model. J. Immunol. 2017, 198, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Hartl, C.A.; Bertschi, A.; Puerto, R.B.; Andresen, C.; Cheney, E.M.; Mittendorf, E.A.; Guerriero, J.L.; Goldberg, M.S. Combination Therapy Targeting Both Innate and Adaptive Immunity Improves Survival in a Pre-Clinical Model of Ovarian Cancer. J. Immunother. Cancer 2019, 7, 199. [Google Scholar] [CrossRef]
- Quaranta, V.; Schmid, M.C. Macrophage-Mediated Subversion of Anti-Tumour Immunity. Cells 2019, 8, 747. [Google Scholar] [CrossRef] [Green Version]
- Kowal, J.; Kornete, M.; Joyce, J.A. Re-Education of Macrophages as a Therapeutic Strategy in Cancer. Immunotherapy 2019, 11, 677–689. [Google Scholar] [CrossRef]
- Buhtoiarov, I.N.; Sondel, P.M.; Eickhoff, J.C.; Rakhmilevich, A.L. Macrophages Are Essential for Antitumour Effects against Weakly Immunogenic Murine Tumours Induced by Class B CpG-Oligodeoxynucleotides. Immunology 2007, 120, 412–423. [Google Scholar] [CrossRef]
- Grzelak, A.; Polakova, I.; Smahelova, J.; Vackova, J.; Pekarcikova, L.; Tachezy, R.; Smahel, M. Experimental Combined Immunotherapy of Tumours with Major Histocompatibility Complex Class I Downregulation. Int. J. Mol. Sci. 2018, 19, 3693. [Google Scholar] [CrossRef] [Green Version]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stangé, G.; van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; de Baetselier, P.; et al. Different Tumor Microenvironments Contain Functionally Distinct Subsets of Macrophages Derived from Ly6C (High) Monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Li, Q.; Qin, L.; Zhao, S.; Wang, J.; Chen, X. Transition of Tumor-Associated Macrophages from MHC Class II (Hi) to MHC Class II (Low) Mediates Tumor Progression in Mice. BMC Immunol. 2011, 12, 43. [Google Scholar] [CrossRef]
- Lum, H.D.; Buhtoiarov, I.N.; Schmidt, B.E.; Berke, G.; Paulnock, D.M.; Sondel, P.M.; Rakhmilevich, A.L. Tumoristatic Effects of Anti-CD40 MAb-Activated Macrophages Involve Nitric Oxide and Tumour Necrosis Factor-Alpha. Immunology 2006, 118, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Ellyard, J.I.; Quah, B.J.C.; Simson, L.; Parish, C.R. Alternatively Activated Macrophage Possess Antitumor Cytotoxicity That Is Induced by IL-4 and Mediated by Arginase-1. J. Immunother. 2010, 33, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages Impede CD8 T Cells from Reaching Tumor Cells and Limit the Efficacy of Anti-PD-1 Treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoreau, M.; Penny, H.L.; Tan, K.; Regnier, F.; Weiss, J.M.; Lee, B.; Johannes, L.; Dransart, E.; Le Bon, A.; Abastado, J.-P.; et al. Vaccine-Induced Tumor Regression Requires a Dynamic Cooperation between T Cells and Myeloid Cells at the Tumor Site. Oncotarget 2015, 6, 27832–27846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, E.R.; Chitu, V. CSF-1 Receptor Signaling in Myeloid Cells. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-Cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Ao, J.-Y.; Zhu, X.-D.; Chai, Z.-T.; Cai, H.; Zhang, Y.-Y.; Zhang, K.-Z.; Kong, L.-Q.; Zhang, N.; Ye, B.-G.; Ma, D.-N.; et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiehagen, K.R.; Girgis, N.M.; Yamada, D.H.; Smith, A.A.; Chan, S.R.; Grewal, I.S.; Quigley, M.; Verona, R.I. Combination of CD40 Agonism and CSF-1R Blockade Reconditions Tumor-Associated Macrophages and Drives Potent Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 1109–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef]
- Lin, K.Y.; Guarnieri, F.G.; Staveley-O’Carroll, K.F.; Levitsky, H.I.; August, J.T.; Pardoll, D.M.; Wu, T.C. Treatment of Established Tumors with a Novel Vaccine That Enhances Major Histocompatibility Class II Presentation of Tumor Antigen. Cancer Res. 1996, 56, 21–26. [Google Scholar] [PubMed]
- Smahel, M.; Síma, P.; Ludvíková, V.; Marinov, I.; Pokorná, D.; Vonka, V. Immunisation with Modified HPV16 E7 Genes against Mouse Oncogenic TC-1 Cell Sublines with Downregulated Expression of MHC Class I Molecules. Vaccine 2003, 21, 1125–1136. [Google Scholar] [CrossRef]
- Lhotakova, K.; Grzelak, A.; Polakova, I.; Vackova, J.; Smahel, M. Establishment and Characterization of a Mouse Tumor Cell Line with Irreversible Downregulation of MHC Class I Molecules. Oncol. Rep. 2019, 42, 2826–2835. [Google Scholar] [CrossRef] [PubMed]
- Smahel, M.; Polakova, I.; Duskova, M.; Ludvikova, V.; Kastankova, I. The Effect of Helper Epitopes and Cellular Localization of an Antigen on the Outcome of Gene Gun DNA Immunization. Gene Ther. 2014, 21, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Smahel, M.; Síma, P.; Ludvíková, V.; Vonka, V. Modified HPV16 E7 Genes as DNA Vaccine against E7-Containing Oncogenic Cells. Virology 2001, 281, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.; Sidney, J.; Southwood, S.; Ruppert, J.; Oseroff, C.; Maewal, A.; Snoke, K.; Serra, H.M.; Kubo, R.T.; Sette, A. Development of High Potency Universal DR-Restricted Helper Epitopes by Modification of High Affinity DR-Blocking Peptides. Immunity 1994, 1, 751–761. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, M.; Charoentong, P.; Hackl, H.; Fischer, M.L.; Snajder, R.; Krogsdam, A.M.; Waldner, M.J.; Bindea, G.; Mlecnik, B.; Galon, J.; et al. Characterization of the Immunophenotypes and Antigenomes of Colorectal Cancers Reveals Distinct Tumor Escape Mechanisms and Novel Targets for Immunotherapy. Genome Biol. 2015, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-Cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A Web Tool for Visualizing Clustering of Multivariate Data Using Principal Component Analysis and Heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Kaštánková, I.; Poláková, I.; Dušková, M.; Šmahel, M. Combined Cancer Immunotherapy Against Aurora Kinase A. J. Immunother. 2016, 39, 160–170. [Google Scholar] [CrossRef]
- Ferrari, M.; Fornasiero, M.C.; Isetta, A.M. MTT Colorimetric Assay for Testing Macrophage Cytotoxic Activity in Vitro. J. Immunol. Methods 1990, 131, 165–172. [Google Scholar] [CrossRef]
- Corraliza, I.M.; Campo, M.L.; Soler, G.; Modolell, M. Determination of Arginase Activity in Macrophages: A Micromethod. J. Immunol. Methods 1994, 174, 231–235. [Google Scholar] [CrossRef]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; de Ruiz-Rosado, J.D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routes, J.M.; Morris, K.; Ellison, M.C.; Ryan, S. Macrophages Kill Human Papillomavirus Type 16 E6-Expressing Tumor Cells by Tumor Necrosis Factor Alpha- and Nitric Oxide-Dependent Mechanisms. J. Virol. 2005, 79, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.I.; Liao, J.C.; Kuo, L. Arginase Modulates Nitric Oxide Production in Activated Macrophages. Am. J. Physiol. 1998, 274, H342–H348. [Google Scholar] [CrossRef]
- Mori, M. Regulation of Nitric Oxide Synthesis and Apoptosis by Arginase and Arginine Recycling. J. Nutr. 2007, 137, 1616S–1620S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, S.; Wang, X.; Liu, P.; Ke, C.; Xu, S. Arginine Metabolism and Deprivation in Cancer Therapy. Biomed. Pharmacother. 2019, 118, 109210. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Reinis, M.; Símová, J.; Bubeník, J. Inhibitory Effects of Unmethylated CpG Oligodeoxynucleotides on MHC Class I-Deficient and -Proficient HPV16-Associated Tumours. Int. J. Cancer 2006, 118, 1836–1842. [Google Scholar] [CrossRef]
- Šmahel, M.; Poláková, I.; Sobotková, E.; Vajdová, E. Systemic Administration of CpG Oligodeoxynucleotide and Levamisole as Adjuvants for Gene-Gun-Delivered Antitumor DNA Vaccines. Clin. Dev. Immunol. 2011, 2011. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Cui, S.; Reichner, J.S.; Mateo, R.B.; Albina, J.E. Activated Murine Macrophages Induce Apoptosis in Tumor Cells through Nitric Oxide-Dependent or -Independent Mechanisms. Cancer Res. 1994, 54, 2462–2467. [Google Scholar]
- Tate, D.J.; Patterson, J.R.; Velasco-Gonzalez, C.; Carroll, E.N.; Trinh, J.; Edwards, D.; Aiyar, A.; Finkel-Jimenez, B.; Zea, A.H. Interferon-Gamma-Induced Nitric Oxide Inhibits the Proliferation of Murine Renal Cell Carcinoma Cells. Int. J. Biol. Sci. 2012, 8, 1109–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahat, M.A.; Hemmerlein, B. Macrophage-Tumor Cell Interactions Regulate the Function of Nitric Oxide. Front. Physiol. 2013, 4, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.-J.; Boshuizen, M.C.S.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [Green Version]
- Mirlekar, B.; Pylayeva-Gupta, Y. IL-12 Family Cytokines in Cancer and Immunotherapy. Cancers 2021, 13, 167. [Google Scholar] [CrossRef] [PubMed]
- Rahim, S.S.; Khan, N.; Boddupalli, C.S.; Hasnain, S.E.; Mukhopadhyay, S. Interleukin-10 (IL-10) Mediated Suppression of IL-12 Production in RAW 264.7 Cells Also Involves c-Rel Transcription Factor. Immunology 2005, 114, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Halder, K.; Ghosh, S.; Bose, A.; Majumdar, S. The Combination of a Novel Immunomodulator with a Regulatory T Cell Suppressing Antibody (DTA-1) Regress Advanced Stage B16F10 Solid Tumor by Repolarizing Tumor Associated Macrophages in Situ. Oncoimmunology 2015, 4, e995559. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, X.; Wu, X.; Wei, M.Q.; Zhang, B.; Liu, X.; Wang, Y. IL-10 Signalling Blockade at the Time of Immunization Inhibits Human Papillomavirus 16 E7 Transformed TC-1 Tumour Cells Growth in Mice. Cell Immunol. 2014, 290, 145–151. [Google Scholar] [CrossRef]
- Bialkowski, L.; van der Jeught, K.; Bevers, S.; Tjok Joe, P.; Renmans, D.; Heirman, C.; Aerts, J.L.; Thielemans, K. Immune Checkpoint Blockade Combined with IL-6 and TGF-β Inhibition Improves the Therapeutic Outcome of MRNA-Based Immunotherapy. Int. J. Cancer 2018, 143, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Li, Y.; Huang, W.; Feng, X.; Sun, P.; Yao, Y.; Yang, X.; Sun, W.; Bai, H.; Liu, C.; et al. Combined Immunization against TGF-Β1 Enhances HPV16 E7-Specific Vaccine-Elicited Antitumour Immunity in Mice with Grafted TC-1 Tumours. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Gyori, D.; Lim, E.L.; Grant, F.M.; Spensberger, D.; Roychoudhuri, R.; Shuttleworth, S.J.; Okkenhaug, K.; Stephens, L.R.; Hawkins, P.T. Compensation between CSF1R+ Macrophages and Foxp3+ Treg Cells Drives Resistance to Tumor Immunotherapy. JCI Insight 2018, 3, e120631. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-Tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeuillard, E.; Yang, J.; Buckarma, E.; Wang, J.; Liu, Y.; Conboy, C.; Pavelko, K.D.; Li, Y.; O’Brien, D.; Wang, C.; et al. Targeting Tumor-Associated Macrophages and Granulocytic Myeloid-Derived Suppressor Cells Augments PD-1 Blockade in Cholangiocarcinoma. J. Clin. Investig. 2020, 130, 5380–5396. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, M.; Andersson, E.; Villabona, L.; Seliger, B.; Lundqvist, A.; Kiessling, R.; Masucci, G.V. HLA-Dependent Tumour Development: A Role for Tumour Associate Macrophages? J. Transl. Med. 2013, 11, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dammeijer, F.; Lievense, L.A.; Kaijen-Lambers, M.E.; van Nimwegen, M.; Bezemer, K.; Hegmans, J.P.; van Hall, T.; Hendriks, R.W.; Aerts, J.G. Depletion of Tumor-Associated Macrophages with a CSF-1R Kinase Inhibitor Enhances Antitumor Immunity and Survival Induced by DC Immunotherapy. Cancer Immunol. Res. 2017, 5, 535–546. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Conjugate | Clone | Company | Staining | Panels | |
|---|---|---|---|---|---|---|
| Arg-1 | APC | A1exF5 | eBioscience | Intracellular | • | |
| CD11b | BV421 | M1/70 | BioLegend | Surface | • | • |
| CD11c | APC-Cy7 | N418 | BioLegend | Surface | • | • |
| CD38 | BV605 | 90 | BD Biosciences | Surface | • | • |
| CD45 | AF700 | 30-F11 | BioLegend | Surface | • | • |
| CSF-1R | PerCP-eFluor710 | AFS98 | eBioscience | Surface | • | |
| Egr2 | PE | erongr2 | eBioscience | Intracellular | • | • |
| F4/80 | BV650 | BM8 | BioLegend | Surface | • | • |
| IDO1 | PerCP-eFluor710 | mIDO-48 | eBioscience | Intracellular | • | |
| iNOS | AF488 | CXNFT | eBioscience | Intracellular | • | |
| Ly6C | BV785 | HK1.4 | BioLegend | Surface | • | • |
| Ly6G | PE-Cy5 | 1A8 | Reagent Genie | Surface | • | • |
| MHC-II | PE-Cy7 | M5/114.15.2 | BioLegend | Surface | • | • |
| TNF-α | PE/Dazzle594 | MP6-XT22 | BioLegend | Intracellular | • | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piatakova, A.; Polakova, I.; Smahelova, J.; Johari, S.D.; Nunvar, J.; Smahel, M. Distinct Responsiveness of Tumor-Associated Macrophages to Immunotherapy of Tumors with Different Mechanisms of Major Histocompatibility Complex Class I Downregulation. Cancers 2021, 13, 3057. https://doi.org/10.3390/cancers13123057
Piatakova A, Polakova I, Smahelova J, Johari SD, Nunvar J, Smahel M. Distinct Responsiveness of Tumor-Associated Macrophages to Immunotherapy of Tumors with Different Mechanisms of Major Histocompatibility Complex Class I Downregulation. Cancers. 2021; 13(12):3057. https://doi.org/10.3390/cancers13123057
Chicago/Turabian StylePiatakova, Adrianna, Ingrid Polakova, Jana Smahelova, Shweta Dilip Johari, Jaroslav Nunvar, and Michal Smahel. 2021. "Distinct Responsiveness of Tumor-Associated Macrophages to Immunotherapy of Tumors with Different Mechanisms of Major Histocompatibility Complex Class I Downregulation" Cancers 13, no. 12: 3057. https://doi.org/10.3390/cancers13123057