
STING Agonists as Cancer Therapeutics
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. STING and Type I Interferons in Anti-Tumor Immunity
Pattern Recognition Receptors
1.2. Role of STING in Driving Immune Responses
1.3. Impact of STING on Tumor Vasculature and Tertiary Lymphoid Structures
2. STING Agonists in Clinical Development
2.1. Cyclic Dinucleotides
2.2. Non-Cyclic Dinucleotides
2.3. Bacterial Vectors
3. STING Agonists in Pre-Clinical Evaluations
3.1. Novel Cyclic Dinucleotides
3.2. Next-Generation Non-Cyclic Dinucleotides
3.3. ENPP1 Inhibitors
3.4. Novel STING Agonists for Systemic Delivery
4. Novel STING Agonist Delivery Platforms
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Shekarian, T.; Valsesia-Wittmann, S.; Brody, J.; Michallet, M.C.; Depil, S.; Caux, C.; Marabelle, A. Pattern recognition receptors: Immune targets to enhance cancer immunotherapy. Ann. Oncol. 2017, 28, 1756–1766. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, Y.; Wang, Y.; Zhang, M.; Sun, W.; Dai, T.; Wang, A.; Wu, X.; Zhang, S.; Wang, S.; et al. A dual role of type I interferons in antitumor immunity. Adv. Biosyst. 2020, 1900237, 1–14. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinchieri, G.; Santoli, D. Anti-viral activity induced by culturing lymphocytes with tumor-derived or virus-transformed cells: Enhancement of human natural killer cell activity by interferon and antagonistic inhibition of susceptibility of target cells to lysis*. J. Exp. Med. 1978. [Google Scholar] [CrossRef]
- Lee, C.-K.; Rao, D.T.; Gertner, R.; Gimeno, R.; Frey, A.B.; Levy, D.E. Distinct requirements for IFNs and STAT1 in NK cell function. J. Immunol. 2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoya, M.; Schiavoni, G.; Mattel, F.; Gresser, I.; Belardelli, F.; Borrow, P.; Tough, D.F. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 2002. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011. [Google Scholar] [CrossRef]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Von Marschall, Z.; Scholz, A.; Cramer, T.; Schäfer, G.; Schirner, M.; Öberg, K.; Wiedenmann, B.; Höcker, M.; Rosewicz, S. Effects of interferon alpha on vascular endothelial growth factor gene transcription and tumor angiogenesis. J. Natl. Cancer Inst. 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ylldlrlm, C.; Nieuwenhuis, S.; Teunissen, P.F.; Horrevoets, A.J.G.; Van Royen, N.; Van Der Pouw Kraan, T.C.T.M. Interferon-beta, a decisive factor in angiogenesis and arteriogenesis. J. Interf. Cytokine Res. 2015, 35, 411–420. [Google Scholar]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type i interferon-dependent innate immunity. Nature 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Montealegre Sanchez, G.A.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 2014. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.L.N.; Pellowe, E.J.; De Jesus, A.A.; Goldbach-Mansky, R.; Hilliard, T.N.; Ramanan, A.V. Interstitial lung disease caused by STING-associated vasculopathy with onset in infancy. Am. J. Respir. Crit. Care Med. 2016, 194, 639–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, H.; Chinn, I.K.; Hong, D.; Orange, J.S.; Lupski, J.R.; Mendoza, A.; Pedroza, L.A.; Barber, G.N. Pro-inflammation associated with a gain-of-function mutation (R284S) in the innate immune sensor STING. Cell Rep. 2018. [Google Scholar] [CrossRef] [Green Version]
- König, N.; Fiehn, C.; Wolf, C.; Schuster, M.; Cura Costa, E.; Tüngler, V.; Alvarez, H.A.; Chara, O.; Engel, K.; Goldbach-Mansky, R.; et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann. Rheum. Dis. 2017. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Kiwerska, K.; Szyfter, K. DNA repair in cancer initiation, progression, and therapy—A double-edged sword. J. Appl. Genet. 2019, 60, 329–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. MLH1 deficiency-triggered DNA hyperexcision by exonuclease 1 activates the cGAS-STING pathway. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Talens, F.; Van Vugt, M.A.T.M. Inflammatory signaling in genomically instable cancers. Cell Cycle 2019, 18, 1830–1848. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Xiao, X.; Yang, X.; Zhang, Z.; Wu, L.; Liu, Z. STING signaling in tumorigenesis and cancer therapy: A friend or foe? Cancer Lett. 2017, 402, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Reisländer, T.; Groelly, F.J.; Tarsounas, M. DNA damage and cancer immunotherapy: A STING in the tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef]
- Campisi, M.; Sundararaman, S.K.; Shelton, S.E.; Knelson, E.H.; Mahadevan, N.R.; Yoshida, R.; Tani, T.; Ivanova, E.; Cañadas, I.; Osaki, T.; et al. Tumor-derived cGAMP regulates activation of the vasculature. Front. Immunol. 2020. [Google Scholar] [CrossRef]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 2019. [Google Scholar] [CrossRef] [Green Version]
- Andzinski, L.; Spanier, J.; Kasnitz, N.; Kröger, A.; Jin, L.; Brinkmann, M.M.; Kalinke, U.; Weiss, S.; Jablonska, J.; Lienenklaus, S. Growing tumors induce a local STING dependent type I IFN response in dendritic cells. Int. J. Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, W.S.; Kong, S.J.; Kim, C.G.; Kim, J.H.; Chang, S.K.; Kim, S.; Kim, G.; Chon, H.J.; Kim, C. STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J. Clin. Invest. 2019. [Google Scholar] [CrossRef] [Green Version]
- Chelvanambi, M.; Fecek, R.J.; Taylor, J.L.; Storkus, W.J. STING agonist-based treatment promotes vascular normalization and tertiary lymphoid structure formation in the therapeutic melanoma microenvironment. J. Immunother. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef] [Green Version]
- Daei Farshchi Adli, A.; Jahanban-Esfahlan, R.; Seidi, K.; Samandari-Rad, S.; Zarghami, N. An overview on Vadimezan (DMXAA): The vascular disrupting agent. Chem. Biol. Drug Des. 2018, 91, 996–1006. [Google Scholar] [CrossRef]
- Baguley, B.C. Antivascular therapy of cancer: DMXAA. Lancet Oncol. 2003, 4, 141–1148. [Google Scholar] [CrossRef]
- Howe, F.A.; McPhail, L.D.; Griffiths, J.R.; McIntyre, D.J.O.; Robinson, S.P. Vessel size index magnetic resonance imaging to monitor the effect of antivascular treatment in a rodent tumor model. Int. J. Radiat. Oncol. Biol. Phys. 2008. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.E.; Hermans, I.F.; Roberts, J.M.; Ching, L.M.; Ronchese, F. 5,6-Dimethylxanthenone-4-acetic acid treatment of a non-immunogenic tumour does not synergize with active or passive CD8+ T-cell immunotherapy. Immunol. Cell Biol. 2006. [Google Scholar] [CrossRef]
- Lemos, H.; Mohamed, E.; Huang, L.; Ou, R.; Pacholczyk, G.; Arbab, A.S.; Munn, D.; Mellor, A.L. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Gulen, M.F.; Koch, U.; Haag, S.M.; Schuler, F.; Apetoh, L.; Villunger, A.; Radtke, F.; Ablasser, A. Signalling strength determines proapoptotic functions of STING. Nat. Commun. 2017. [Google Scholar] [CrossRef]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.D.; Seano, G.; Jain, R.K. Normalizing function of tumor vessels: Progress, opportunities, and challenges. Annu. Rev. Physiol. 2019, 81, 505–534. [Google Scholar] [CrossRef]
- Bose, A.; Taylor, J.L.; Alber, S.; Watkins, S.C.; Garcia, J.A.; Rini, B.I.; Ko, J.S.; Cohen, P.A.; Finke, J.H.; Storkus, W.J. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int. J. Cancer 2011. [Google Scholar] [CrossRef] [PubMed]
- Downey, C.M.; Aghaei, M.; Schwendener, R.A.; Jirik, F.R. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2′3′-cGAMP, induces M2 macrophage repolarization. PLoS ONE 2014. [Google Scholar] [CrossRef] [Green Version]
- Demaria, O.; De Gassart, A.; Coso, S.; Gestermann, N.; Di Domizio, J.; Flatz, L.; Gaide, O.; Michielin, O.; Hwu, P.; Petrova, T.V.; et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipi, E.; Nayar, S.; Gardner, D.H.; Colafrancesco, S.; Smith, C.; Barone, F. Tertiary lymphoid structures: Autoimmunity goes local. Front. Immunol. 2018, 9, 1952. [Google Scholar] [CrossRef] [PubMed]
- Dieu-Nosjean, M.C.; Goc, J.; Giraldo, N.A.; Sautès-Fridman, C.; Fridman, W.H. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014, 35, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Neyt, K.; Perros, F.; GeurtsvanKessel, C.H.; Hammad, H.; Lambrecht, B.N. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. 2012, 33, 297–305. [Google Scholar] [CrossRef]
- Weinstein, A.M.; Storkus, W.J. Biosynthesis and functional significance of peripheral node addressin in cancer-associated TLO. Front. Immunol. 2016, 7, 301. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.; Gallimore, A.; Ager, A. Defining high endothelial venules and tertiary lymphoid structures in cancer. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2018. [Google Scholar]
- Denton, A.E.; Innocentin, S.; Carr, E.J.; Bradford, B.M.; Lafouresse, F.; Mabbott, N.A.; Mörbe, U.; Ludewig, B.; Groom, J.R.; Good-Jacobson, K.L.; et al. Type I interferon induces CXCL13 to support ectopic germinal center formation. J. Exp. Med. 2019. [Google Scholar] [CrossRef] [Green Version]
- Cufi, P.; Dragin, N.; Ruhlmann, N.; Weiss, J.M.; Fadel, E.; Serraf, A.; Berrih-Aknin, S.; Le Panse, R. Central role of interferon-beta in thymic events leading to myasthenia gravis. J. Autoimmun. 2014. [Google Scholar] [CrossRef]
- Martinet, L.; Le Guellec, S.; Filleron, T.; Lamant, L.; Meyer, N.; Rochaix, P.; Garrido, I.; Girard, J.P. High endothelial venules (HEVs) in human melanoma lesions: Major gateways for tumor-infiltrating lymphocytes. Oncoimmunology 2012. [Google Scholar] [CrossRef] [Green Version]
- Kuerten, S.; Schickel, A.; Kerkloh, C.; Recks, M.S.; Addicks, K.; Ruddle, N.H.; Lehmann, P.V. Tertiary lymphoid organ development coincides with determinant spreading of the myelin-specific T cell response. Acta Neuropathol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, K.; Wang, S.; Sagan, S.A.; Zamvil, S.S.; von Büdingen, H.-C. B cell repertoire expansion occurs in meningeal ectopic lymphoid tissue. JCI Insight 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwabara, S.; Tsuchikawa, T.; Nakamura, T.; Hatanaka, Y.; Hatanaka, K.C.; Sasaki, K.; Ono, M.; Umemoto, K.; Suzuki, T.; Sato, O.; et al. Prognostic relevance of tertiary lymphoid organs following neoadjuvant chemoradiotherapy in pancreatic ductal adenocarcinoma. Cancer Sci. 2019. [Google Scholar] [CrossRef]
- Sakimura, C.; Tanaka, H.; Okuno, T.; Hiramatsu, S.; Muguruma, K.; Hirakawa, K.; Wanibuchi, H.; Ohira, M. B cells in tertiary lymphoid structures are associated with favorable prognosis in gastric cancer. J. Surg. Res. 2017. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Avram, G.; Sánchez-Sendra, B.; Martín, J.M.; Terrádez, L.; Ramos, D.; Monteagudo, C. The density and type of MECA-79-positive high endothelial venules correlate with lymphocytic infiltration and tumour regression in primary cutaneous melanoma. Histopathology 2013. [Google Scholar] [CrossRef]
- Messina, J.L.; Fenstermacher, D.A.; Eschrich, S.; Qu, X.; Berglund, A.E.; Lloyd, M.C.; Schell, M.J.; Sondak, V.K.; Weber, J.S.; Mulé, J.J. 12-chemokine gene signature identifies lymph node-like structures in melanoma: Potential for patient selection for immunotherapy? Sci. Rep. 2012. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Su, H.; Xu, D.; Dai, W.; Zhang, W.; Wang, Z.; Anderson, C.F.; Zheng, M.; Oh, R.; Wan, F.; et al. Tumour sensitization via the extended intratumoural release of a STING agonist and camptothecin from a self-assembled hydrogel. Nat. Biomed. Eng. 2020. [Google Scholar] [CrossRef]
- Junkins, R.D.; Gallovic, M.D.; Johnson, B.M.; Collier, M.A.; Watkins-Schulz, R.; Cheng, N.; David, C.N.; McGee, C.E.; Sempowski, G.D.; Shterev, I.; et al. A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J. Control. Release 2018. [Google Scholar] [CrossRef]
- McKeage, M.J.; Von Pawel, J.; Reck, M.; Jameson, M.B.; Rosenthal, M.A.; Sullivan, R.; Gibbs, D.; Mainwaring, P.N.; Serke, M.; Lafitte, J.J.; et al. Randomised phase II study of ASA404 combined with carboplatin and paclitaxel in previously untreated advanced non-small cell lung cancer. Br. J. Cancer 2008. [Google Scholar] [CrossRef] [PubMed]
- Lara, P.N.; Douillard, J.Y.; Nakagawa, K.; Von Pawel, J.; McKeage, M.J.; Albert, I.; Losonczy, G.; Reck, M.; Heo, D.S.; Fan, X.; et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J. Clin. Oncol. 2011. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwar, J.R.; Kanwar, R.K.; Pandey, S.; Ching, L.M.; Krissansen, G.W. Vascular attack by 5,6-dimethylxanthenone-4-acetic acid combined with B7.1 (CD80)-mediated immunotherapy overcomes immune resistance and leads to the eradication of large tumors and multiple tumor foci. Cancer Res. 2001, 61, 1948–1956. [Google Scholar] [PubMed]
- Shih, A.Y.; Damm-Ganamet, K.L.; Mirzadegan, T. Dynamic structural differences between human and mouse STING lead to differing sensitivity to DMXAA. Biophys. J. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivick, K.E.; Desbien, A.L.; Glickman, L.H.; Reiner, G.L.; Corrales, L.; Surh, N.H.; Hudson, T.E.; Vu, U.T.; Francica, B.J.; Banda, T.; et al. Magnitude of therapeutic STING activation determines CD8+ T cell-mediated anti-tumor immunity. Cell Rep. 2018. [Google Scholar] [CrossRef] [Green Version]
- Francica, B.J.; Ghasemzadeh, A.; Desbien, A.L.; Theodros, D.; Sivick, K.E.; Reiner, G.L.; Glickman, L.H.; Marciscano, A.E.; Sharabi, A.B.; Leong, M.L.; et al. TNFa and radioresistant stromal cells are essential for therapeutic efficacy of cyclic dinucleotide STING agonists in nonimmunogenic tumors. Cancer Immunol. Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Foote, J.B.; Kok, M.; Leatherman, J.M.; Armstrong, T.D.; Marcinkowski, B.C.; Ojalvo, L.S.; Kanne, D.B.; Jaffee, E.M.; Dubensky, T.W.; Emens, L.A. A STING agonist given with OX40 receptor and PD-L1 modulators primes immunity and reduces tumor growth in tolerized mice. Cancer Immunol. Res. 2017. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Desbien, A.L.; Gauthier, K.S.; Reiner, G.; Corrales, L.; Schroeder, T.; Glickman, L.H.; Surh, N.H.; Francica, B.; Leong, J.J.; et al. Abstract P351: ADU-S100 (MIW815) synergizes with checkpoint inhibition to elicit an anti-tumor CD8+ T cell response to control distal tumors. In Proceedings of the Society for Immunotherapy of Cancer 33rd Annual Meeting, Washington, DC, USA, 7–11 November 2018. [Google Scholar]
- Meric-Bernstam, F.; Sandhu, S.K.; Hamid, O.; Spreafico, A.; Kasper, S.; Dummer, R.; Shimizu, T.; Steeghs, N.; Lewis, N.; Talluto, C.C.; et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Harrington, K.J.; Brody, J.; Ingham, M.; Strauss, J.; Cemerski, S.; Wang, M.; Tse, A.; Khilnani, A.; Marabelle, A.; Golan, T. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann. Oncol. 2018. [Google Scholar] [CrossRef]
- Challa, S.V.; Zhou, S.; Sheri, A.; Padmanabhan, S.; Meher, G.; Gimi, R.; Schmidt, D.; Cleary, D.; Afdhal, N.; Iyer, R. Preclinical studies of SB 11285, a novel STING agonist for immuno-oncology. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Schieven, G.; Brown, J.; Swanson, J.; Stromko, C.; Ho, C.-P.; Zhang, R.; Li-Wang, B.; Qiu, H.; Sun, H.; Fink, B.; et al. Abstract P525: Preclinical characterization of BMS-986301, a differentiated STING agonist with robust antitumor activity as monotherapy or in combination with anti-PD-1. In Proceedings of the Society for Immunotherapy of Cancer 33rd Annual Meeting, Washington, DC, USA, 7–11 November 2018. [Google Scholar]
- Gremel, G.; Impagnatiello, M.A.; Carotta, S.; Schaaf, O.; Chetta, P.M.; Oost, T.; Zichner, T.; Hofmann, M.; Blake, S.; Bretschneider, T.; et al. Abstract 4522: Potent induction of a tumor-specific immune response by a cyclic dinucleotide STING agonist. Am. Assoc. Cancer Res. 2020, 80, 4522. [Google Scholar]
- ENDO, A.; Kim, D.-S.; Huang, K.-C.; Hao, M.-H.; Mathieu, S.; Choi, H.; Majumder, U.; Zhu, X.; Shen, Y.; Sanders, K.; et al. Abstract 4456: Discovery of E7766: A representative of a novel class of macrocycle-bridged STING agonists (MBSAs) with superior potency and pan-genotypic activity. Am. Assoc. Cancer Res. 2019, 79, 4456. [Google Scholar]
- Huang, K.-C.; Endo, A.; McGrath, S.; Chandra, D.; Wu, J.; Kim, D.-S.; Albu, D.; Ingersoll, C.; Tendyke, K.; Loiacono, K.; et al. Abstract 3269: Discovery and characterization of E7766, a novel macrocycle-bridged STING agonist with pan-genotypic and potent antitumor activity through intravesical and intratumoral administration. Am. Assoc. Cancer Res. 2019, 79, 3269. [Google Scholar]
- Huang, K.-C.; Zhang, C.; Yu, K.; Kim, D.-S.; Dixit, V.; Hukkanen, R.; Choi, H.-W.; Hutz, J.; Fang, F.; Bao, X. Abstract 592: Demonstration of E7766, a novel STING agonist, as a potent immunotherapy in BCG-insensitive non-muscle invasive bladder cancer models via intravesical administration. Am. Assoc. Cancer Res. 2020, 80, 592. [Google Scholar]
- Leventhal, D.S.; Sokolovska, A.; Li, N.; Plescia, C.; Kolodziej, S.A.; Gallant, C.W.; Christmas, R.; Gao, J.R.; James, M.J.; Abin-Fuentes, A.; et al. Immunotherapy with engineered bacteria by targeting the STING pathway for anti-tumor immunity. Nat. Commun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Glickman, L.H.; Skoble, J.; Rae, C.S.; Makarova, A.M.; D’Antonio, M.A.; McGeehan, A.J.; Thanos, C. Abstract P235: STACT-TREX1: A novel tumor-targeting systemically-delivered STING pathway agonist demonstrates robust anti-tumor efficacy in multiple murine cancer models. In Proceedings of the Society for Immunotherapy of Cancer 33rd Annual Meeting, Washington, DC, USA, 7–11 November 2018. [Google Scholar]
- Makarova, A.M.; Iannello, A.; Rae, C.S.; King, B.; Besprozvannaya, M.; Faulhaber, J.; Skoble, J.; Thanos, C.D.; Glickman, L.H. Abstract 5016: STACT-TREX1: A systemically-administered STING pathway agonist targets tumor-resident myeloid cells and induces adaptive anti-tumor immunity in multiple preclinical models. Am. Assoc. Cancer Res. 2019, 79, 5016. [Google Scholar]
- Smith, M.; Chin, D.; Chan, S.; Mahady, S.; Campion, L.; Morgan, C.; Patel, S.; Chu, G.; Hughes, A.; Bignan, G.; et al. Abstract 5567: In vivo administration of the STING agonist, JNJ-67544412, leads to complete regression of established murine subcutaneous tumors. Am. Assoc. Cancer Res. 2020, 80, 5567. [Google Scholar]
- Thomsen, M.K. Abstract 2344: The cGAS-STING pathway is a therapeutic target in a preclinical model of hepatocellular carcinoma. Oncogene 2019, 39, 1652–1664. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Adam, M.; Clemens, J.; Creech, K.; Schneck, J.; Pasikanti, K.; Tran, J.-L.; Joglekar, D.; Hopson, C.; Pesiridis, S.; et al. Abstract 5554: Preclinical characterization of GSK532, a novel STING agonist with potent anti-tumor activity. Am. Assoc. Cancer Res. 2018, 78, 5554. [Google Scholar]
- Chmielewski, S.; Zawadzka, M.; Mazurek, J.; Rogacki, M.K.; Gluza, K.; Wójcik-Jaszczyńska, K.; Poczkaj, A.; Ćwiertnia, G.; Topolnicki, G.; Kujawa, M.; et al. Abstract 4532A: Development of selective small molecule STING agonists suitable for systemic administration. Am. Assoc. Cancer Res. 2020, 80, 5432A. [Google Scholar]
- Binder, G.A.; Gambino, C.S.; Kharitonova, A.; Metcalf, R.S.; Daniel, K.G.; Guida, W.C. Abstract 6: Computationally assisted target screening of STING agonist for immunologic therapy. Am. Assoc. Cancer Res. 2019, 79, 6. [Google Scholar]
- Dobrzańska, M.; Chmielewski, S.; Zawadzka, M.; Mazurek, J.; Gluza, K.; Wójcik-Jaszczyńska, K.; Kujawa, M.; Topolnicki, G.; Ćwiertnia, G.; Poczkaj, A.; et al. Abstract 4983: Discovery and characterization of next-generation small molecule direct STING agonists. Am. Assoc. Cancer Res. 2019, 79, 4983. [Google Scholar]
- Wang, Z.; Dove, P.; Rosa, D.; Bossen, B.; Helke, S.; Charbonneau, M.; Brinen, L.; Dodge, K.; Lin, G.H.; Galligan, C.; et al. Abstract 3854: Preclinical characterization of a novel non-cyclic dinucleotide small molecule STING agonist with potent antitumor activity in mice. Am. Assoc. Cancer Res. 2019, 79, 3854. [Google Scholar]
- Chan, S.R.; Bignan, G.; Pierson, E.; Mahady, S.; Ta, H.; Schepens, W.; Thuring, J.W.; Lim, H.K.; Otieno, M.; Wilde, T.; et al. Abstract 5567A: JNJ-‘6196: A next generation STING agonist with potent preclinical activity by the IV route. Am. Assoc. Cancer Res. 2020, 80, 5567A. [Google Scholar]
- Banerjee, M.; Basu, S.; Middya, S.; Shrivastava, R.; Ghosh, R.; Pryde, D.C.; Yadav, D.; Bhattacharya, G.; Soram, T.; Puniya, K.; et al. Abstract LB-061: CRD5500: A versatile small molecule STING agonist amenable to bioconjugation as an ADC. Am. Assoc. Cancer Res. 2019, 79, LB-061. [Google Scholar]
- Li, A.; Song, Y.; Dong, C.; Chen, X.; Yang, J. Abstract 3317: Discovery of novel STING agonists with robust anti-tumor activity. Am. Assoc. Cancer Res. 2020, 80, 3317. [Google Scholar]
- Perera, S.A.; Kopinja, J.E.; Ma, Y.; Laskey, J.; Chakravarthy, K.; Chen, Y.; Cui, L.; Presland, J.; Zhao, S.; Minnihan, E.; et al. Abstract 4721: Combining STING agonists with an anti-PD-1 antagonist results in marked antitumor activity in immune-excluded tumors. Am. Assoc. Cancer Res. 2018, 78, 4721. [Google Scholar]
- Jekle, A.; Thatikonda, S.; Stevens, S.; Williams, C.; Kinkade, A.; Ren, S.; Jaisinghani, R.; Zhang, Q.; Misner, D.; Stoycheva, A.; et al. Abstract 4520: Preclinical characterization of ALG-031048, a novel STING agonist with potent anti-tumor activity in mice. Am. Assoc. Cancer Res. 2020, 80, 4520. [Google Scholar]
- Weston, A.S.; Thode, T.G.; Rodriguez del Villar, R.; Dana, S.; Kasibhatla, S.; Kaadige, M.R.; Sharma, S. Abstract LB-118: SR8541A is a potent inhibitor of ENPP1 and exhibits dendritic cell mediated antitumor activity. Cancer Res. 2020, 80 (Suppl. 16). [Google Scholar] [CrossRef]
- Weston, A.; Thode, T.; Munoz, R.; Daniel, S.; Soldi, R.; Kaadige, M.; Han, H.; Vankayalapti, H.; Sharma, S. Abstract 3077: Preclinical studies of SR-8314, a highly selective ENPP1 inhibitor and an activator of STING pathway. Am. Assoc. Cancer Res. 2019, 79, 3077. [Google Scholar]
- Sharma, S.; Weston, A.; Thode, T.; Gomez, E.; Kaadige, M.; Vankayalapti, H. Abstract 1932: Discovery of ENPP1 inhibitors as agonists of STING pathway. Am. Assoc. Cancer Res. 2018, 78, 1932. [Google Scholar]
- Baird, J.; Dietsch, G.; Florio, V.; Gallatin, M.; Knox, C. MV-626, a potent and selective inhibitor of ENPP1 enhances STING activation and augments T-cell mediated anti-tumor activity in vivo. In Proceedings of the Poster presented at Society for Immunotherapy of Cancer Annual Meeting, Washington, DC, USA, 7–11 November 2018; Available online: https://digitalcommons.psjhealth.org/sitc2018/7 (accessed on 29 May 2021).
- Bukhalid, R.A.; Duvall, J.R.; Cetinbas, N.M.; Catcott, K.C.; Avocetien, K.; Bentley, K.W.; Bradley, S.; Carter, T.; Chin, C.-N.; Clardy, S.; et al. Abstract 6706: Systemic Administration of STING Agonist Antibody-Drug Conjugates Elicit Potent Anti-Tumor Immune Responses with Minimal Induction of Circulating Cytokines. Cancer Res. 2020, 80 (Suppl. 16). [Google Scholar] [CrossRef]
- Miller, J.; Luo, M.; Wang, H.; Wang, Z.; Ding, X.; Campbell, A.; Almazan, J.; Gutowski, S.; Chen, Z.; Gao, J.; et al. Abstract 4577: ONM-500: A STING-activating therapeutic nanovaccine platform for cancer immunotherapy. Am. Assoc. Cancer Res. 2020, 80, 4577. [Google Scholar]
- Mi, Y.; Smith, C.C.; Serody, J.S.; Vincent, B.G.; Wang, A.Z.; Hyun, H. Abstract 2866: Neoantigen nanovaccine improves personalized cancer immunotherapy. Am. Assoc. Cancer Res. 2020, 80, 2866. [Google Scholar]
- Jang, S.C.; Moniz, R.J.; Sia, C.L.; Harrison, R.A.; Houde, D.; Ross, N.; Xu, K.; Lewis, N.; Bourdeau, R.; McCoy, C.; et al. Abstract 944: ExoSTING: An engineered exosome therapeutic that selectively delivers STING agonist to the tumor resident antigen-presenting cells resulting in improved tumor antigen-specific adaptive immune response. Am. Assoc. Cancer Res. 2019, 79, 94. [Google Scholar]
- Jang, S.C.; Moniz, R.J.; Sia, C.; Dey, J. Abstract P618: Selective delivery of exosome-mediated STING agonist to antigen presenting cells results in significantly improved potency and reduced toxicity. In Proceedings of the Society for Immunotherapy of Cancer 33rd Annual Meeting, Washington, DC, USA, 7–11 November 2018. [Google Scholar]
- Cheng, N.; Watkins-Schulz, R.; Junkins, R.; David, C.; Johnson, B.M.; Montgomery, S.A.; Peine, K.J.; Darr, D.B.; Yuan, H.; McKinnon, K.P.; et al. Abstract LB-126: Nanoparticle-incorporated STING activator as an immunotherapeutic for PD-L1 resistant triple-negative breast cancer. Am. Assoc. Cancer Res. 2018, 78, LB-126. [Google Scholar]
- Thomsen, M.K.; Skouboe, M.K.; Boularan, C.; Vernejoul, F.; Lioux, T.; Leknes, S.L.; Berthelsen, M.F.; Riedel, M.; Cai, H.; Joseph, J.V.; et al. The cGAS-STING pathway is a therapeutic target in a preclinical model of hepatocellular carcinoma. Oncogene 2020. [Google Scholar] [CrossRef]
- Jekle, A.; Thatikonda, S.; Jaisinghani, R.; Ren, S. Tumor regression in a mouse model of hepatocellular carcinoma upon treatment with the STING agonist ALG-031048. In Proceedings of the American Association for the Study of Liver Diseases Annual Meeting; 2020. (virtual). Available online: https://www.aligos.com/wp-content/uploads/2020/11/Tumor-regression-in-a-mouse-model-of-hepatocellular-carcinoma-upon-treatment-with-the-STING-agonist-ALG-031048.pdf (accessed on 29 May 2021).
- Onyedibe, K.I.; Wang, M.; Sintim, H.O. ENPP1, an old enzyme with new functions, and small molecule inhibitors—A sting in the tale of ENPP1. Molecules 2019, 24, 4192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, K.; Nishimasu, H.; Oikawa, D.; Hirano, S.; Hirano, H.; Kasuya, G.; Ishitani, R.; Tokunaga, F.; Nureki, O. Structural insights into cGAMP degradation by Ecto-nucleotide pyrophosphatase phosphodiesterase 1. Nat. Commun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yin, Q.; Kuss, P.; Maliga, Z.; Millán, J.L.; Wu, H.; Mitchison, T.J. Hydrolysis of 2’3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.Y.; Tran, J.L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.N.; Yu, C.; Vartabedian, V.F.; Jia, Y.; Kumar, M.; Gamo, A.M.; Vernier, W.; Ali, S.H.; Kissai, M.; Lazar, D.C.; et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 2020. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.S.; Perera, S.A.; Piesvaux, J.A.; Presland, J.P.; Schroeder, G.K.; Cumming, J.N.; Wesley Trotter, B.; Altman, M.D.; Buevich, A.V.; Cash, B.; et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, S.; Wang, X.Y.; Zhu, G. Nanovaccines for cancer immunotherapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnology 2019, 11, 1–29. [Google Scholar] [CrossRef]
- Luo, M.; Wang, H.; Wang, Z.; Cai, H.; Lu, Z.; Li, Y.; Du, M.; Huang, G.; Wang, C.; Chen, X.; et al. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 2017. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wang, L.; Freywald, A.; Qureshi, M.; Chen, Y.; Xiang, J. A novel T cell-based vaccine capable of stimulating long-term functional CTL memory against B16 melanoma via CD40L signaling. Cell. Mol. Immunol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Erb, U.; Zhao, K.; Hackert, T.; Zöller, M. Efficacy of vaccination with tumor-exosome loaded dendritic cells combined with cytotoxic drug treatment in pancreatic cancer. Oncoimmunology 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, B.; Ilyukha, V.; Sorokin, M.; Buzdin, A.; Vannier, E.; Poltorak, A. Cutting edge: Activation of STING in T cells induces type I IFN responses and cell death. J. Immunol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Lemos, H.; Ou, R.; McCardle, C.; Lin, Y.; Calver, J.; Minett, J.; Chadli, A.; Huang, L.; Mellor, A.L. Overcoming resistance to STING agonist therapy to incite durable protective antitumor immunity. J. Immunother. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.T.; Shae, D.; Gonzalez-Ericsson, P.I.; Sanchez, V.; Gong, J.; Liang, Y.; Hinerfeld, D.; Beechem, J.M.; Balko, J.M. Abstract 4978: Digital spatial profiling of molecular responses to nanoparticle STING agonists identify S100A9 and B7-H3 as possible escape mechanisms. Am. Assoc. Cancer Res. 2019, 79, 4978. [Google Scholar]
- Konno, H.; Yamauchi, S.; Berglund, A.; Putney, R.M.; Mulé, J.J.; Barber, G.N. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]


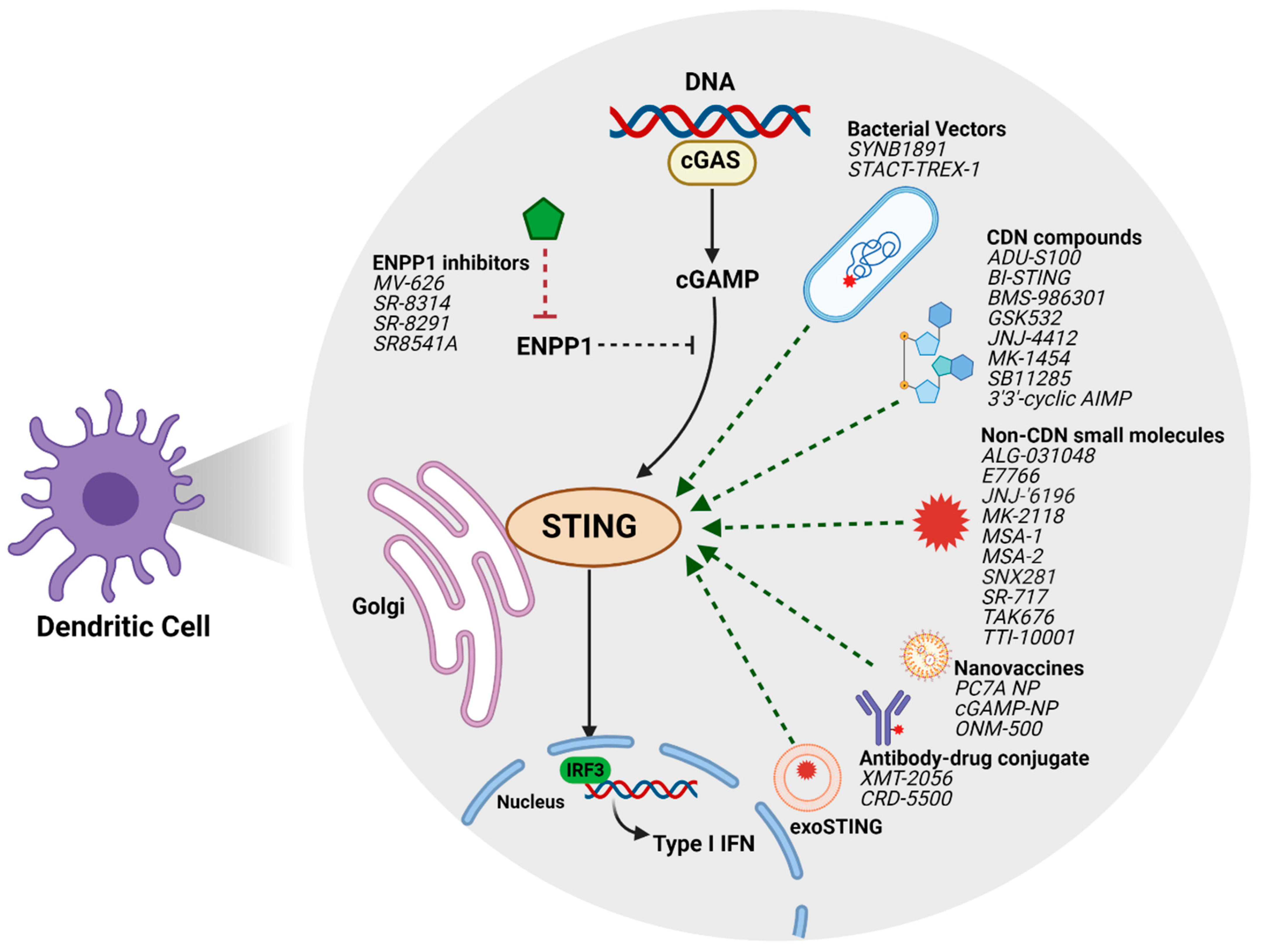{kind=link}
| Agent | Route of Delivery | Phase | Type of Cancer | Clinical Trial NCT Code | |
|---|---|---|---|---|---|
| ADU-S100/MIW815 | Single agent or + Ipilimumab | IT | Phase I | Advanced/Metastatic Solid Tumors or Lymphomas | NCT02675439 |
| + Pembrolizumab | IT | Phase II | PD-L1 positive recurrent or metastatic HNSCC | NCT03937141 | |
| + PDR001 | IT | Phase Ib | Advanced/Metastatic Solid Tumors or Lymphomas | NCT03172936 | |
| MK-1454 | Single agent or + Pembrolizumab | IT | Phase I | Advanced/Metastatic Solid Tumors or Lymphomas | NCT03010176 |
| + Pembrolizumab | IT | Phase II | Metastatic or Unresectable, Recurrent HNSCC | NCT04220866 | |
| MK-2118 | + Pembrolizumab | IT/SQ | Phase I | Advanced/Metastatic Solid Tumors or Lymphomas | NCT03249792 |
| SB11285 | Single agent or + Atezolizumab | IV | Phase Ia/Ib | Advanced Solid Tumors | NCT04096638 |
| GSK3745417 | Single agent or + Pembrolizumab | IV | Phase I | Advanced Solid Tumors | NCT03843359 |
| BMS-986301 | Single agent or + Nivolumab/ Ipilimumab | IT/IM | Phase I | Advanced Solid Tumors | NCT03956680 |
| BI-STING (BI 1387446) | Single agent or + BI 754091 (anti-PD1 monoclonal antibody) | IT | Phase I | Advanced Solid Tumors | NCT04147234 |
| E7766 | Single agent | IT | Phase I/Ib | Advanced Solid Tumors or Lymphomas | NCT04144140 |
| TAK-676 | Single agent or + Pembrolizumab | IV | Phase I | Advanced Solid Tumors | NCT04420884 |
| SNX281 | Single agent or + Pembrolizumab | IV | Phase I | Advanced Solid Tumors or Lymphomas | NCT04609579 |
| SYNB1891 | Single agent or + Atezolizumab | IT | Phase I | Advanced Solid Tumors or Lymphomas | NCT04167137 |
| Agent | Structure/Properties | Route of Delivery | Tumor Model | Findings | References |
|---|---|---|---|---|---|
| Cyclic dinucleotide (CDN) | |||||
| JNJ-67544412 (JNJ-4412) | Cyclic dinucleotide, Potently binds to all major human STING alleles | Intratumoral | Subcutaneous syngeneic murine tumor models |
| [84] |
| BI-STING | Mimics natural STING ligand | Intratumoral | Subcutaneous syngeneic murine tumor models |
| [77] |
| 3′3′-cyclic 3′3′-cAIMP | Cyclic dinucleotide | Not specified | Mouse model of mutagen-induced hepatocellular carcinoma |
| [85] |
| GSK532 | Cyclic dinucleotide | Intratumoral | CT26 murine syngeneic model |
| [86] |
| Non-CDN Agonists | |||||
| Ryvu’s agonists | Selective non-nucleotide, non-macrocyclic, small molecule compounds, potential for systemic administration | Not specified | CT26 murine syngeneic model |
| [87] |
| GF3-002 | Novel low-molecular-weight organic molecule, not based on a CDN | In vitro | In vitro assays |
| [88] |
| Selvita agonists | Selective non-nucleotide, non-macrocyclic, small molecule compounds, structurally unrelated to known CDNs, tunable properties with enhanced plasma stability and permeability, potential for systemic administration | In vitro | In vitro assays |
| [89] |
| TTI-10001 | Non-CDN small molecule STING agonist | Intratumoral | Multiple syngeneic murine tumor models |
| [90] |
| JNJ-‘6196 | Next-generation STING agonist; binds to STING with weaker affinity and a faster off rate, but more potent than other CDNs in activating dendritic cells | Intravenous | Murine tumor models (not specified) |
| [91] |
| CRD5500 | Next-generation small molecule STING agonist. Activates all five common human STING variants. Delivery via different routes (IV or SC) or as an antibody drug conjugate | Intravenous, subcutaneous, Antibody-drug conjugate (ADC) with Trastuzumab | CT26 syngeneic murine model |
| [92] |
| CS-1018, CS-1020 and CS-1010 | STING agonists with higher potency in activating mouse and human STING variants than natural ligand cGAMP | Intratumoral | B16F10 and MC38 murine tumor models |
| [93] |
| MSA-1 | Novel STING agonist with higher potency in activating STING protein than cGAMP | Intratumoral | MC38 syngeneic tumors, CT26 and B16-F10 tumor models |
| [94] |
| ALG-031048 | Novel STING agonist with high potency and superior stability | Intratumoral, Subcutaneous | Syngeneic CT26 colorectal, B16F10 melanoma, and Hepa1–6 HCC models |
| [95] |
| Macrocyclic STING Agonist | |||||
| E7766 | Macrocyclic STING agonist with superior in vitro activity against all major human STING genotypes, chemical and metabolic stability, conferred by conformational rigidity of the unique macrocycle bridge | Intravesical, Intratumoral | Murine anti-PD1 insensitive NMIBC tumor models, subcutaneous tumor models |
| [78,80] |
| ENPP1 Inhibitor | |||||
| SR-8541A | Small molecule ENPP1 inhibitor | In vitro | In vitro assays |
| [96] |
| SR-8314 | Analog of SR-8291 (a highly selective ENPP1 inhibitor) | Intraperitoneal | Syngeneic murine tumor model |
| [97] |
| Orally available ENPP1 inhibitors | Small molecule compounds with strong binding affinity towards ENPP1 | In vitro | In vitro assays |
| [98] |
| MV-626 | Selective ENPP1 inhibitor with 100% oral bioavailability | Intraperitoneal | Panc02-SIY and MC38 murine tumor models |
| [99] |
| Novel Delivery Systems | |||||
| Antibody drug conjugates (ADC) | STING agonist ADCs | Intravenous | Multiple xenograft and syngeneic murine models |
| [100] |
| ONM-500 nanovaccine | Novel pH-sensitive polymer that forms an antigen-encapsulating nanoparticle and functions both as a carrier for antigen delivery to DCs and as an adjuvant, activating the STING pathway | Subcutaneous | TC-1 cervical cancer murine model |
| [101] |
| Neoantigen nanovaccine | Redox-responsive neoantigen-polymer conjugates and a STING agonist DMXAA | Subcutaneous | B16-F10 melanoma murine model |
| [102] |
| exoSTING | Engineered exosome therapeutic that delivers STING agonist to tumor resident APCs | Intratumoral | Checkpoint refractory B16-F10 melanoma murine model |
| [103,104] |
| STACT-TREX1 | Inhibitory microRNA to TREX1, introduced into the STACT strain. | Intravenous | CT26 and MC38 colon carcinoma models, and B16-F10 melanoma model |
| [82,83] |
| STING-NPs | Liposomal nanoparticles (NPs) to deliver the STING agonist, cGAMP | Intravenous | Basal-like triple-negative breast cancer (TNBC) murine model |
| [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. https://doi.org/10.3390/cancers13112695
Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ, Luke JJ. STING Agonists as Cancer Therapeutics. Cancers. 2021; 13(11):2695. https://doi.org/10.3390/cancers13112695
Chicago/Turabian StyleAmouzegar, Afsaneh, Manoj Chelvanambi, Jessica N. Filderman, Walter J. Storkus, and Jason J. Luke. 2021. "STING Agonists as Cancer Therapeutics" Cancers 13, no. 11: 2695. https://doi.org/10.3390/cancers13112695