Immunogenomic Gene Signature of Cell-Death Associated Genes with Prognostic Implications in Lung Cancer


 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Gene Expression Analysis to Determine Cell Death Index (CDI)
2.2. Clinico-Pathological Analysis
2.3. Cox-Proportional Hazard
2.4. Differential Expression of Genes and Principal Component Analysis (PCA)
2.5. Immune Cell Infiltration Analysis
2.6. Evaluation of Cytokines, Checkpoint Molecules and T Cell Exhaustion Genes
2.7. Functional Enrichment Analysis
2.8. Statistical Analysis
3. Results
3.1. Survival Analysis of Patients Using Cell Death Index (CDI)
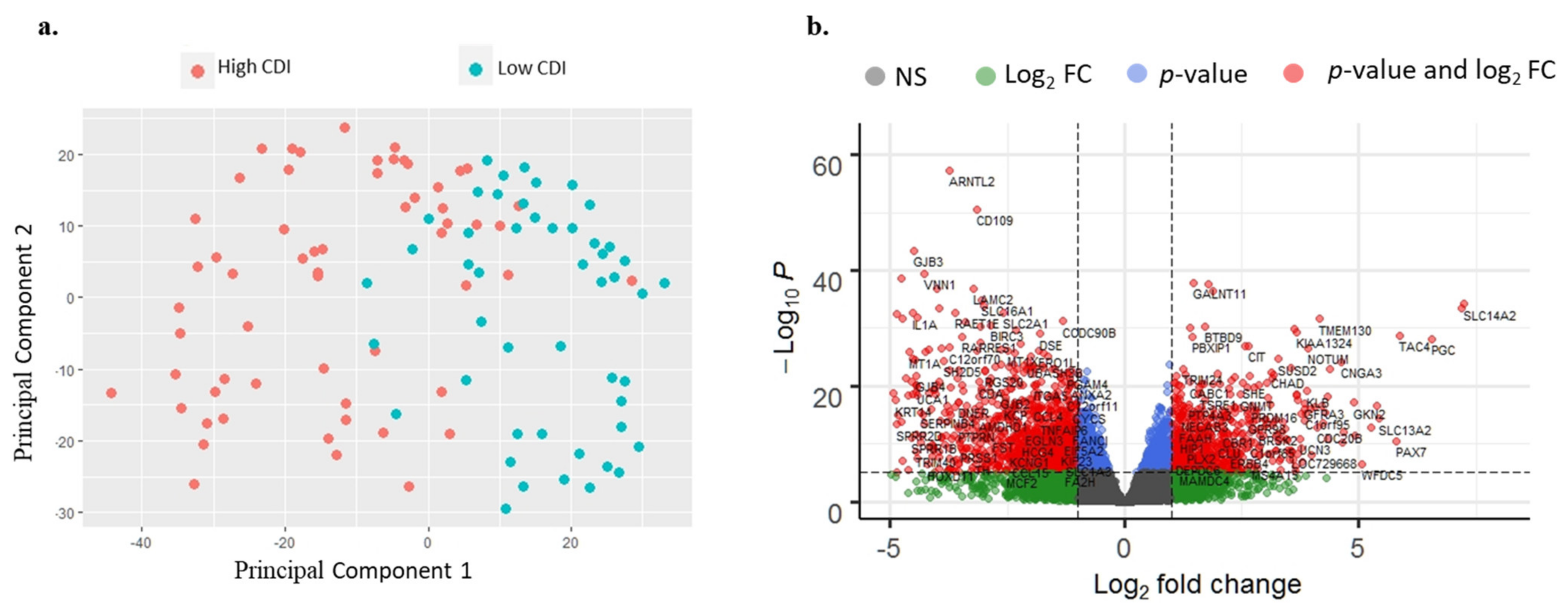
3.2. RNA-Seq Analysis of Patients
3.3. Clinico-Pathological and Survival Analysis
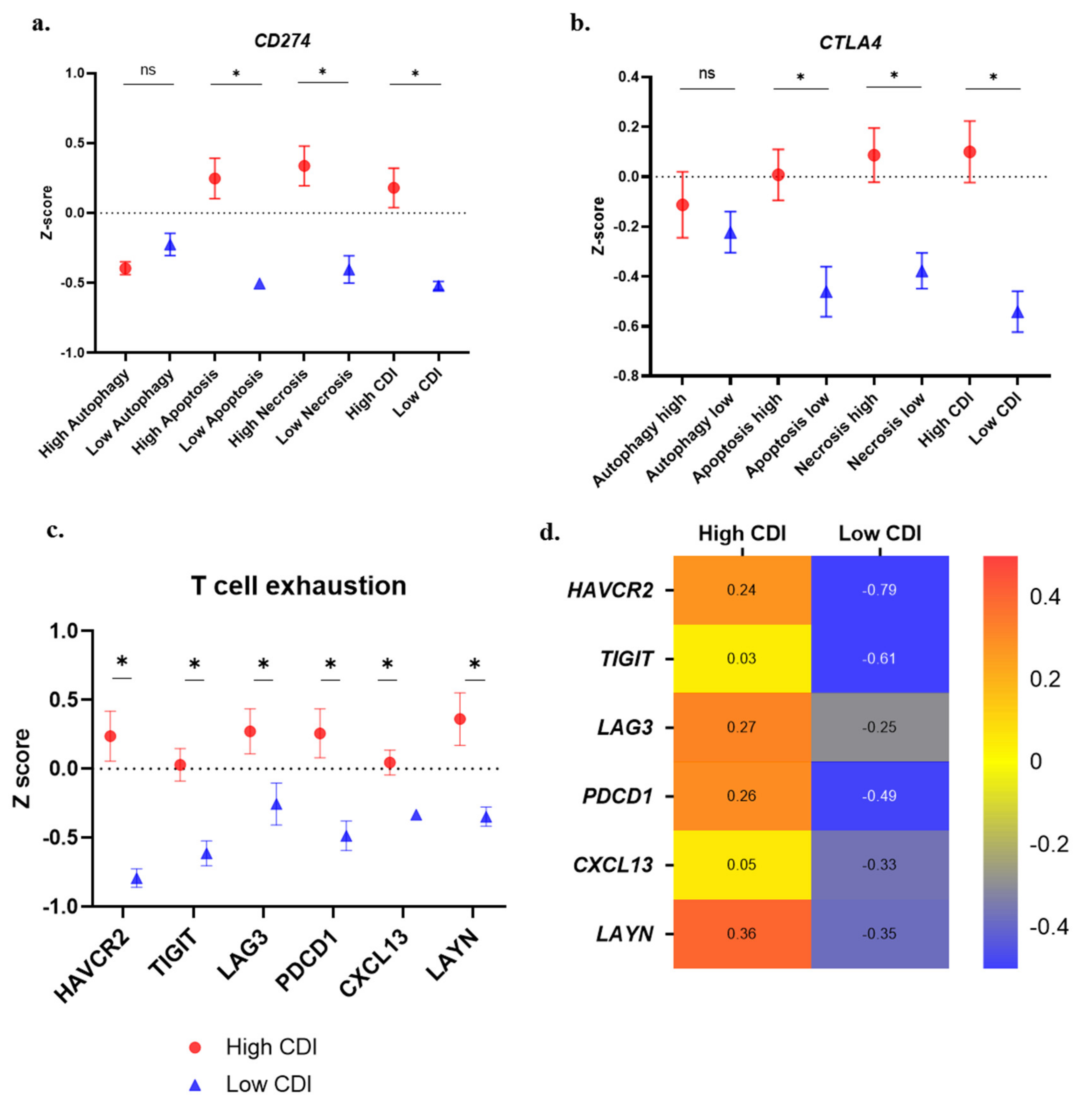
3.4. Cytokine Gene Expression Analysis
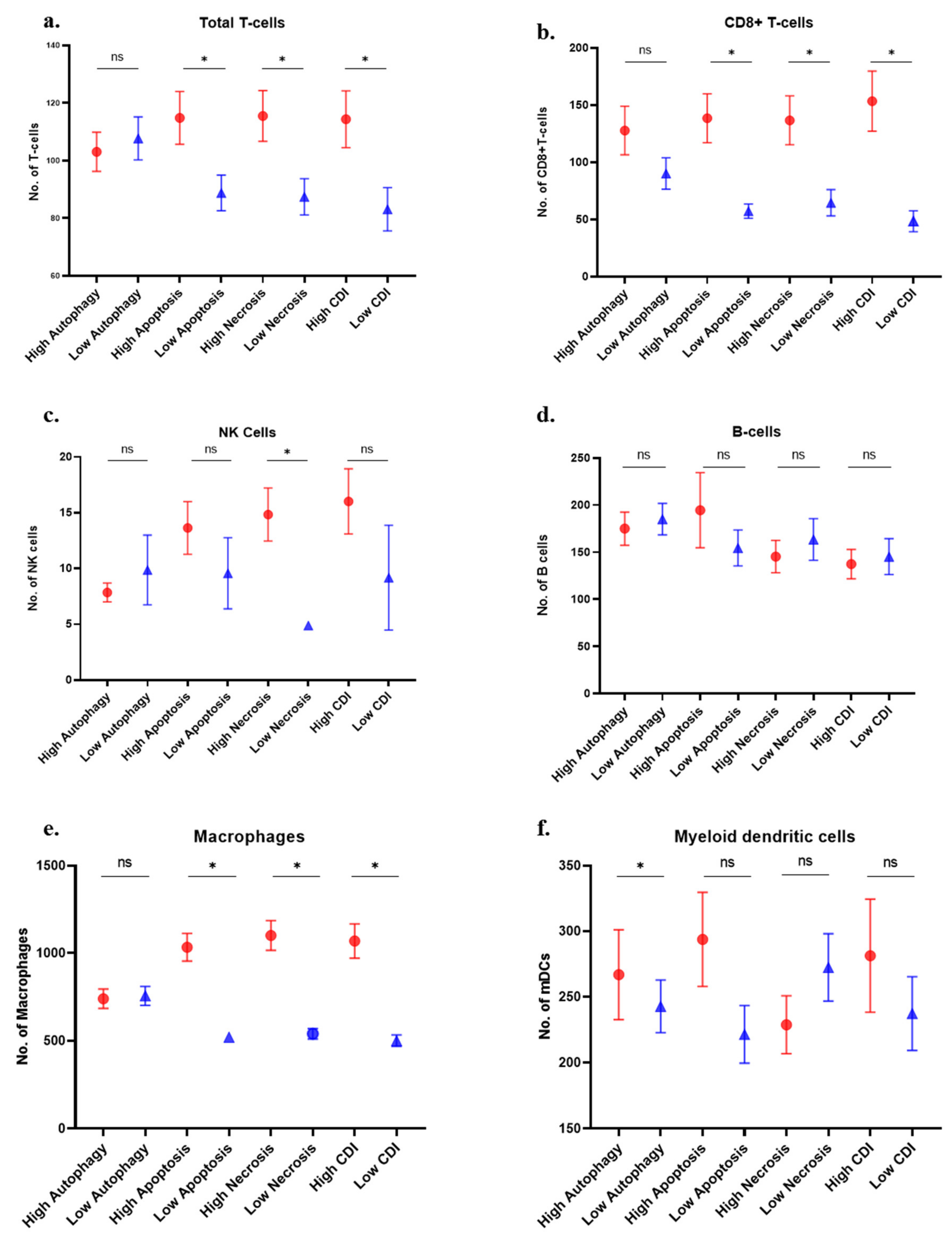
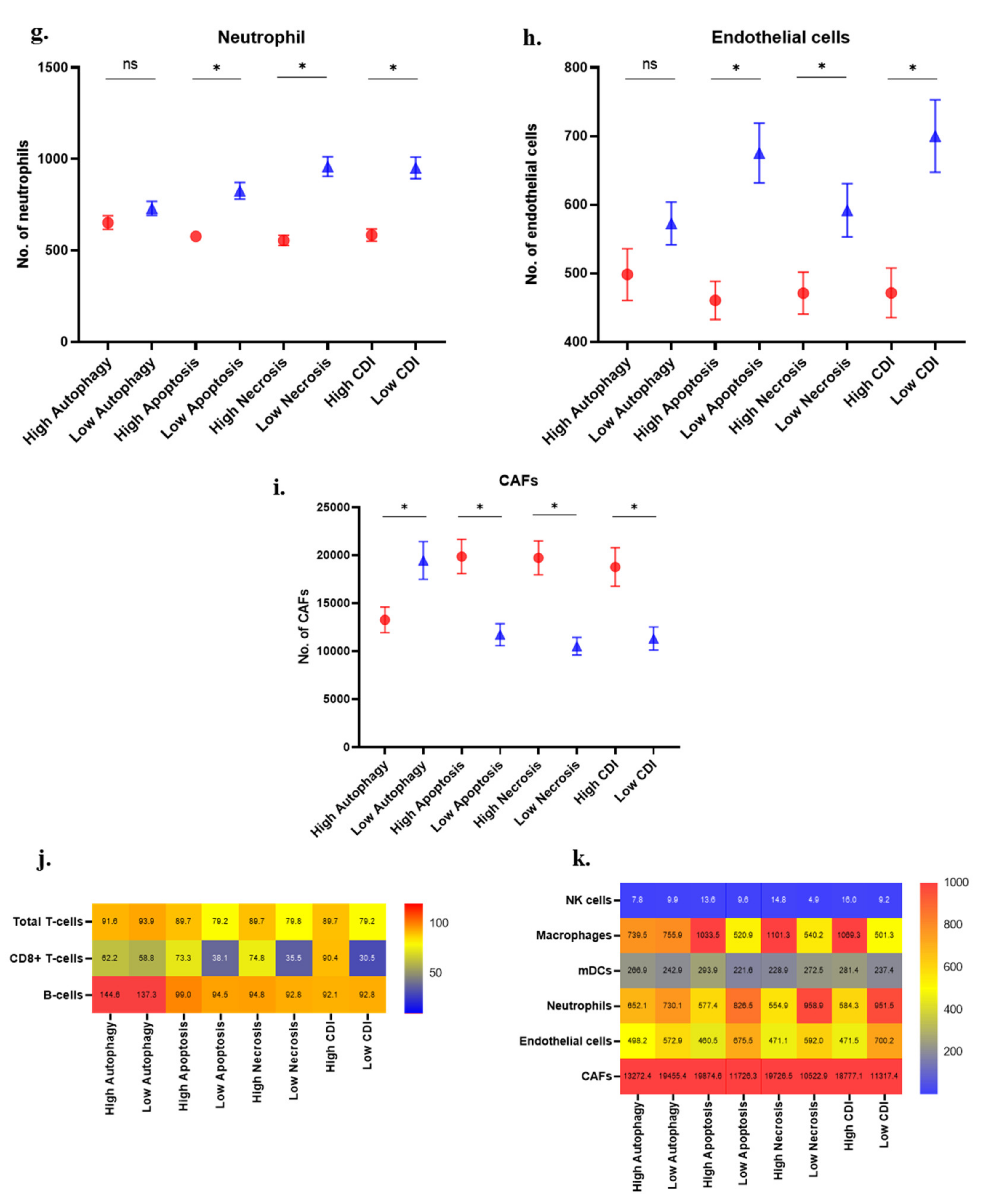
3.5. Immune Cell Analysis
3.6. Immune Suppression and T-Cell Exhaustion Markers
3.7. Enrichment Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Barrta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global Epidemiology of Lung Cancer. Ann. Glob. Health 2019, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Prim. 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Wang, M.; Shen, Z.; Zhu, J. A new immune signature for survival prediction and immune checkpoint molecules in lung adenocarcinoma. J. Transl. Med. 2020, 18, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arneth, B. Tumor Microenvironment. Medicina (Kaunas) 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Ruiz, M.E.; Vitale, I.; Harrington, K.J.; Melero, I.; Galluzzi, L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat. Immunol. 2020, 21, 120–134. [Google Scholar] [CrossRef]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Humeau, J.; Bezu, L.; Kepp, O.; Kroemer, G. EIF2alpha phosphorylation: A hallmark of both autophagy and immunogenic cell death. Mol. Cell. Oncol. 2020, 7, 1776570. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Jan, R.; Chaudhry, G.E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Karsch-Bluman, A.; Feiglin, A.; Arbib, E.; Stern, T.; Shoval, H.; Schwob, O.; Berger, M.; Benny, O. Tissue necrosis and its role in cancer progression. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Yuan, J. Expansion and evolution of cell death programmes. Nat. Rev. Mol. Cell Biol. 2008, 9, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Domagala-Kulawik, J. The role of the immune system in non-small cell lung carcinoma and potential for therapeutic intervention. Transl. Lung Cancer Res. 2015, 4, 177–190. [Google Scholar]
- Aisner, D.L.; Marshall, C.B. Molecular pathology of non-small cell lung cancer: A practical guide. Am. J. Clin. Pathol. 2012, 138, 332–346. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.; Jabbour, S.K.; Aisner, J. Current state of immunotherapy for non-small cell lung cancer. Transl. Lung Cancer Res. 2007, 6, 196–211. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.M.; Stinchcombe, T.E. Second-Line Therapy for Advanced NSCLC. Oncologist 2013, 18, 947–953. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; Zhao, C.; Ozarina, M.; Zhao, X.; Yang, J.; Chen, H. Targeting Immune Checkpoints in Lung Cancer: Current Landscape and Future Prospects. Clin. Drug Investig. 2019, 39, 341–353. [Google Scholar] [CrossRef]
- Guo, N.L.; Wan, Y.W.; Bose, S.; Denvir, J.; Kashon, M.L.; Andrew, M.E. A novel network model identified a 13-gene lung cancer prognostic signature. Int. J. Comput. Biol. Drug Des. 2011, 4, 19–39. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, G.; Sun, N.; Zhang, Z.; Zhang, Z.; Luo, Y.; Che, Y.; Xue, Q.; He, J. Comprehensive molecular analyses of a TNF family-based signature with regard to prognosis, immune features, and biomarkers for immunotherapy in lung adenocarcinoma. EBioMedicine 2020, 59, 102959. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Shen-Orr, S.S.; Gaujoux, R. Computational deconvolution: Extracting cell type-specific information from heterogeneous samples. Curr. Opin. Immunol. 2013, 25, 571–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 1–20. [Google Scholar] [CrossRef]
- Petitprez, F.; Vano, Y.A.; Becht, E.; Giraldo, N.A.; de Reynies, A.; Sautes-Fridman, C.; Fridman, W.H. Transcriptomic analysis of the tumor microenvironment to guide prognosis and immunotherapies. Cancer Immunol. Immunother. 2017, 67, 981–988. [Google Scholar] [CrossRef]
- Chen, C.H.; Lu, T.P. Utilizing gene expression profiles to characterize tumor infiltrating lymphocytes in cancers. Ann. Transl. Med. 2019, 7, S289. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, H.; Mao, B.; Zhou, Y.; Shi, X.; Tang, L.; Jiang, H.; Wang, G.; Zhuang, W. Transcriptional Characterization Of The Tumor Immune Microenvironment And Its Prognostic Value For Locally Advanced Lung Adenocarcinoma in A Chinese Population. Cancer Manag. Res. 2019, 11, 9165–9173. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Deng, X.; Chen, X.; Chen, S.; Song, L.; Meng, M.; Han, Q.; Imani, S.; Li, S.; Zhong, Z.; et al. Landscape of active enhancers developed de novo in cirrhosis and conserved in hepatocellular carcinoma. Am. J. Cancer Res. 2020, 10, 3157–3178. [Google Scholar]
- Yang, H.; Sun, L.; Guan, A.; Yin, H.; Liu, M.; Mao, X.; Xu, H.; Zhao, H.; Lu, X.; Sang, X.; et al. Unique TP53 neoantigen and the immune microenvironment in long-term survivors of Hepatocellular carcinoma. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Song, Q.; Yang, Z.; Sun, X.; Xue, M.; Chen, W.; Yang, J.; Wang, S. Analysis of PD-1 related immune transcriptional profile in different cancer types. Cancer Cell Int. 2018, 18, 218. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Kitsou, M.; Ayiomamitis, G.D.; Zaravinos, A. High expression of immune checkpoints is associated with the TIL load, mutation rate and patient survival in colorectal cancer. Int. J. Oncol. 2020, 57, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [Green Version]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Gkogkou, C.; Frangia, K.; Saif, M.W.; Trigidou, R.; Syrigos, K. Necrosis and apoptotic index as prognostic factors in non-small cell lung carcinoma: A review. Springerplus 2014, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, Q.; Gao, Z.; Xin, S.; Zhao, Y.; Zhang, K.; Shi, R.; Bao, X. A novel 4-gene signature for overall survival prediction in lung adenocarcinoma patients with lymph node metastasis. Cancer Cell Int. 2019, 19, 1–9. [Google Scholar] [CrossRef]
- Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; Seshan, V.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [CrossRef]
- Chen, H.Y.; Yu, S.L.; Chen, C.H.; Chang, G.C.; Chen, C.Y.; Yuan, A.; Cheng, C.L.; Wang, C.H.; Terng, H.J.; Kao, S.F.; et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N. Engl. J. Med. 2007, 356, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, D.P.; Gandara, D.R.; Antonia, S.J.; Zielinski, C.; Paz-Ares, L. Non-Small-Cell Lung Cancer: Role of the Immune System and Potential for Immunotherapy. J. Thorac. Oncol. 2015, 10, 974–984. [Google Scholar] [CrossRef] [Green Version]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, S.; Gong, D.; Qin, Y.; Shen, Q. Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell. Mol. Immunol. 2010, 7, 389–395. [Google Scholar] [CrossRef]
- Chen, Y.B.; Mu, C.Y.; Huang, J.A. Clinical significance of programmed death-1 ligand-1 expression in patients with non-small cell lung cancer: A 5-year-follow-up study. Tumori 2012, 98, 751–755. [Google Scholar] [CrossRef]
- Mu, C.Y.; Huang, J.A.; Chen, Y.; Chen, C.; Zhang, X.G. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med. Oncol. 2011, 28, 682–688. [Google Scholar] [CrossRef]
- Ruffini, E.; Asioli, S.; Filosso, P.L.; Lyberis, P.; Bruna, M.C.; Macri, L.; Daniele, L.; Oliaro, A. Clinical significance of tumor-infiltrating lymphocytes in lung neoplasms. Ann. Thorac. Surg. 2009, 87, 365–372. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkholder, B.; Huang, R.Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.Q.; Gao, C.Y.; Wang, B.L.; Zhang, Y.M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 2014, 1845, 182–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhu, B.; Zhang, M.; Wang, X. Roles of immune microenvironment heterogeneity in therapy-associated biomarkers in lung cancer. Semin. Cell Dev. Biol. 2017, 64, 90–97. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Death Process | Gene | Gene ID | Gene (Full Name) |
|---|---|---|---|
| Autophagy | PRKAA2 | 5563 | Protein Kinase AMP-Activated Catalytic Subunit Alpha 2 |
| ATG12 | 9140 | Autophagy Related 12 | |
| ULK2 | 9706 | Unc-51 Like Autophagy Activating Kinase 2 | |
| ATG5 | 9474 | Autophagy Related 5 | |
| GABARAPL1 | 23710 | GABA Type A Receptor Associated Protein Like 1 | |
| Apoptosis | BCL2L1 | 598 | B-Cell Lymphoma 2 Like 1 |
| CASP9 | 842 | Caspase 9 | |
| CYCS | 54205 | C, Somatic | |
| IL1A | 3552 | Interleukin 1 Alpha | |
| PIK3CG | 5294 | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Gamma | |
| TNFRSF10D | 8793 | Tumor Necrosis Factor Receptor Superfamily Member 10D | |
| FADD | 8772 | Fas Associated Via Death Domain | |
| Apoptosis and Necrosis | BIRC3 | 330 | Baculoviral IAP Repeat-Containing Protein 3 |
| FAS | 355 | Fas Cell Surface Death Receptor | |
| Necrosis | DNM1L | 10059 | Dynamin 1 Like |
| GSDME | 1687 | Gasdermin E | |
| IPMK | 253430 | Inositol Polyphosphate Multikinase | |
| MLKL | 197259 | Mixed Lineage Kinase Domain Like Pseudokinase | |
| RBCK1 | 10616 | RANBP2-Type and C3HC4-Type Zinc Finger Containing 1 | |
| TICAM1 | 148022 | Toll Like Receptor Adaptor Molecule 1 | |
| YBX3 | 8531 | Y-Box Binding Protein 3 |
| (a) | |||||||
|---|---|---|---|---|---|---|---|
| Clinical Variable | High CDI (n = 301) | Low CDI (n = 209) | Pearson χ2 p-Value | ||||
| Age | |||||||
| <66 years | 137 | 101 | 0.63 | ||||
| >66 years | 151 | 102 | |||||
| Ethnicity | |||||||
| African American | 29 | 23 | 0.72 | ||||
| Caucasian | 224 | 160 | |||||
| Sex | |||||||
| Female | 154 | 120 | 0.16 | ||||
| Male | 147 | 89 | |||||
| Stage | |||||||
| I + II | 225 | 173 | 0.01 | ||||
| II + III | 76 | 34 | |||||
| Lymph Node Involvement | |||||||
| N0 | 174 | 154 | 0.01 | ||||
| N1 + N2 + N3 | 122 | 49 | |||||
| Distant Metastasis | |||||||
| No Metastasis M0 | 214 | 127 | 0.78 | ||||
| Metastasis M1 | 15 | 10 | |||||
| Aneuploidy Score | |||||||
| <16 | 127 | 114 | 0.01 | ||||
| >16 | 166 | 90 | |||||
| Fraction Genome Altered | |||||||
| <0.23 | 135 | 107 | 0.22 | ||||
| >0.23 | 156 | 99 | |||||
| Overall Survival (OS) | |||||||
| <5 years | 267 | 177 | 0.06 | ||||
| >5 years | 25 | 28 | |||||
| OS Status | |||||||
| Living | 174 | 151 | 0.01 | ||||
| Deceased | 127 | 58 | |||||
| Progression-Free Survival (PFS) | |||||||
| <5 years | 277 | 186 | 0.06 | ||||
| >5 years | 17 | 21 | |||||
| PFS Status | |||||||
| No progression | 165 | 139 | 0.01 | ||||
| Progression | 136 | 70 | |||||
| Disease-Free Survival (DFS) | |||||||
| <5 Years | 139 | 121 | 0.39 | ||||
| >5 Years | 17 | 20 | |||||
| DFS Status | |||||||
| Disease-free | 102 | 111 | 0.01 | ||||
| Recurred/progressed | 56 | 31 | |||||
| Disease-Specific Survival (DSS) | |||||||
| <5 years | 267 | 177 | 0.06 | ||||
| >5 years | 25 | 28 | |||||
| DSS Status | |||||||
| Tumor free | 194 | 167 | 0.01 | ||||
| Dead with tumor | 84 | 29 | |||||
| (b) | |||||||
| Clinical Variable | Univariate | Multivariate | |||||
| Hazard Ratio | 95% CI | p-Value | Hazard Ratio | 95% CI | p-Value | ||
| Cell death index (CDI High, CDI low) | 1.75 | 1.28–2.45 | <0.001 | 1.62 | 1.11–2.36 | <0.01 | |
| T, Stage (III + IV, I + II) | 2.68 | 1.95–3.63 | <0.001 | 2.13 | 1.39–3.25 | <0.001 | |
| N, Lymph Node involvement (N1 + N2 + N3, N0) | 2.61 | 1.94–3.52 | <0.001 | 2.25 | 1.59–3.19 | <0.001 | |
| M, Distant Metastasis (M1, M0) | 2.12 | 1.19–3.52 | <0.01 | 1.68 | 0.93–3.03 | 0.07 | |
| Age (>66, <66 years) | 1.2 | 0.89–1.62 | 0.21 | ||||
| Sex (Male, Female) | 1.05 | 0.71–0.78 | 0.71 | ||||
| Radiation therapy (Yes, No) | 2.02 | 1.36–2.90 | <0.001 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahluwalia, P.; Ahluwalia, M.; Mondal, A.K.; Sahajpal, N.; Kota, V.; Rojiani, M.V.; Rojiani, A.M.; Kolhe, R. Immunogenomic Gene Signature of Cell-Death Associated Genes with Prognostic Implications in Lung Cancer. Cancers 2021, 13, 155. https://doi.org/10.3390/cancers13010155
Ahluwalia P, Ahluwalia M, Mondal AK, Sahajpal N, Kota V, Rojiani MV, Rojiani AM, Kolhe R. Immunogenomic Gene Signature of Cell-Death Associated Genes with Prognostic Implications in Lung Cancer. Cancers. 2021; 13(1):155. https://doi.org/10.3390/cancers13010155
Chicago/Turabian StyleAhluwalia, Pankaj, Meenakshi Ahluwalia, Ashis K. Mondal, Nikhil Sahajpal, Vamsi Kota, Mumtaz V. Rojiani, Amyn M. Rojiani, and Ravindra Kolhe. 2021. "Immunogenomic Gene Signature of Cell-Death Associated Genes with Prognostic Implications in Lung Cancer" Cancers 13, no. 1: 155. https://doi.org/10.3390/cancers13010155