The Role of Antigen Processing and Presentation in Cancer and the Efficacy of Immune Checkpoint Inhibitor Immunotherapy



Abstract
:Simple Summary
Abstract
1. The Immune System and Cancer
2. Mechanisms of Cancer Immune Evasion and the Role of Immune Checkpoints
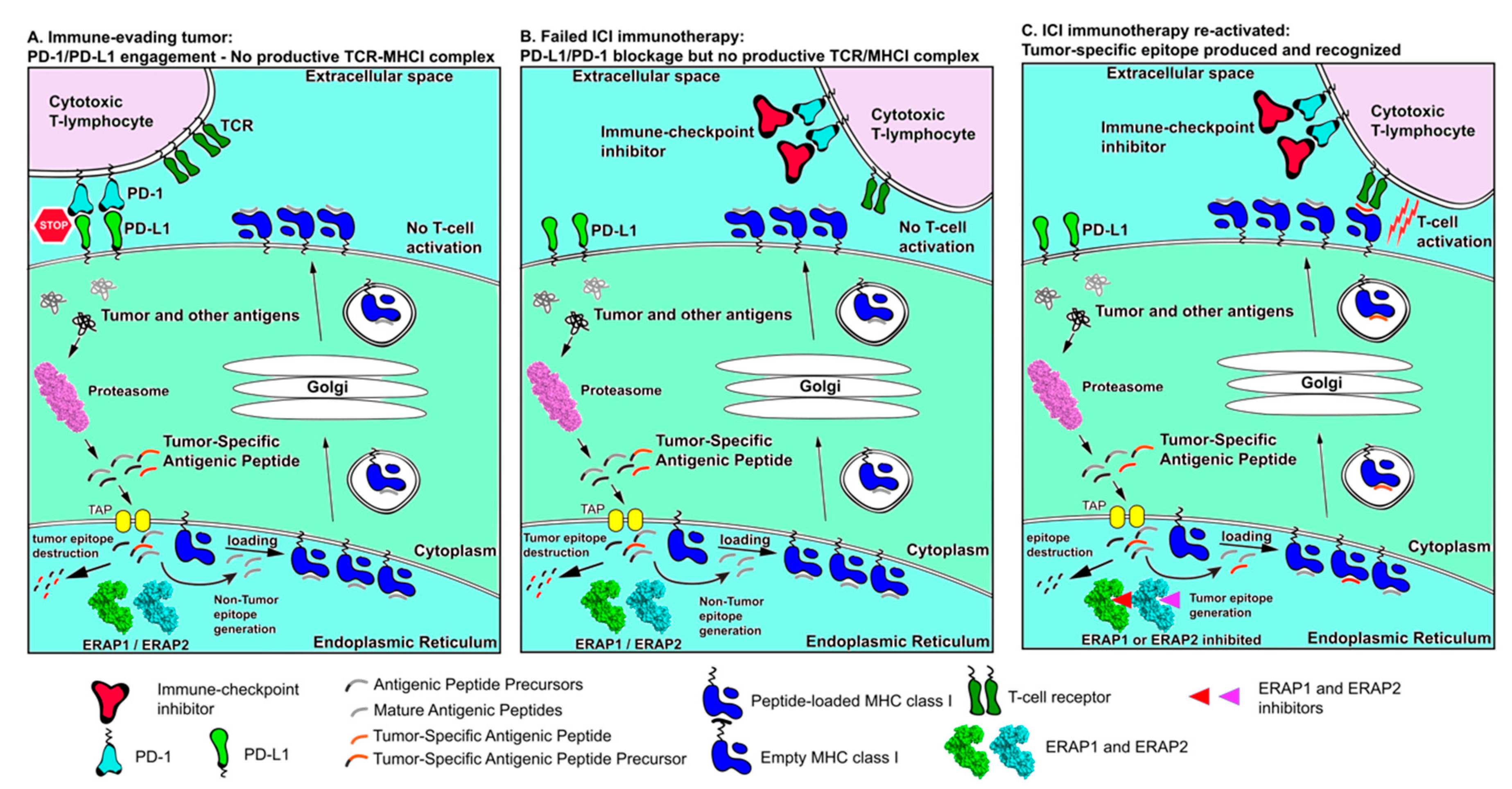
3. ICI Therapy Failure and Tumor Immunogenicity
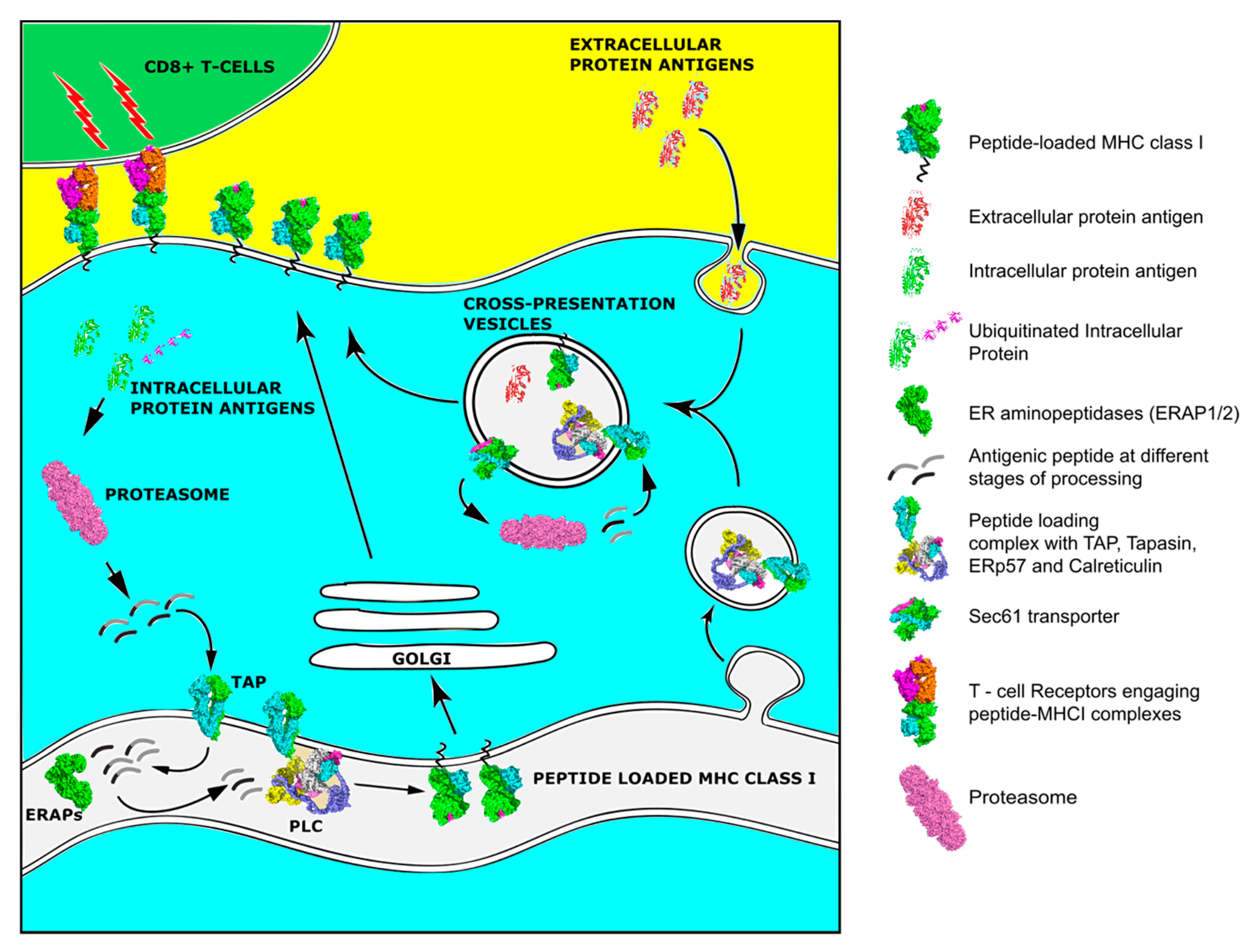
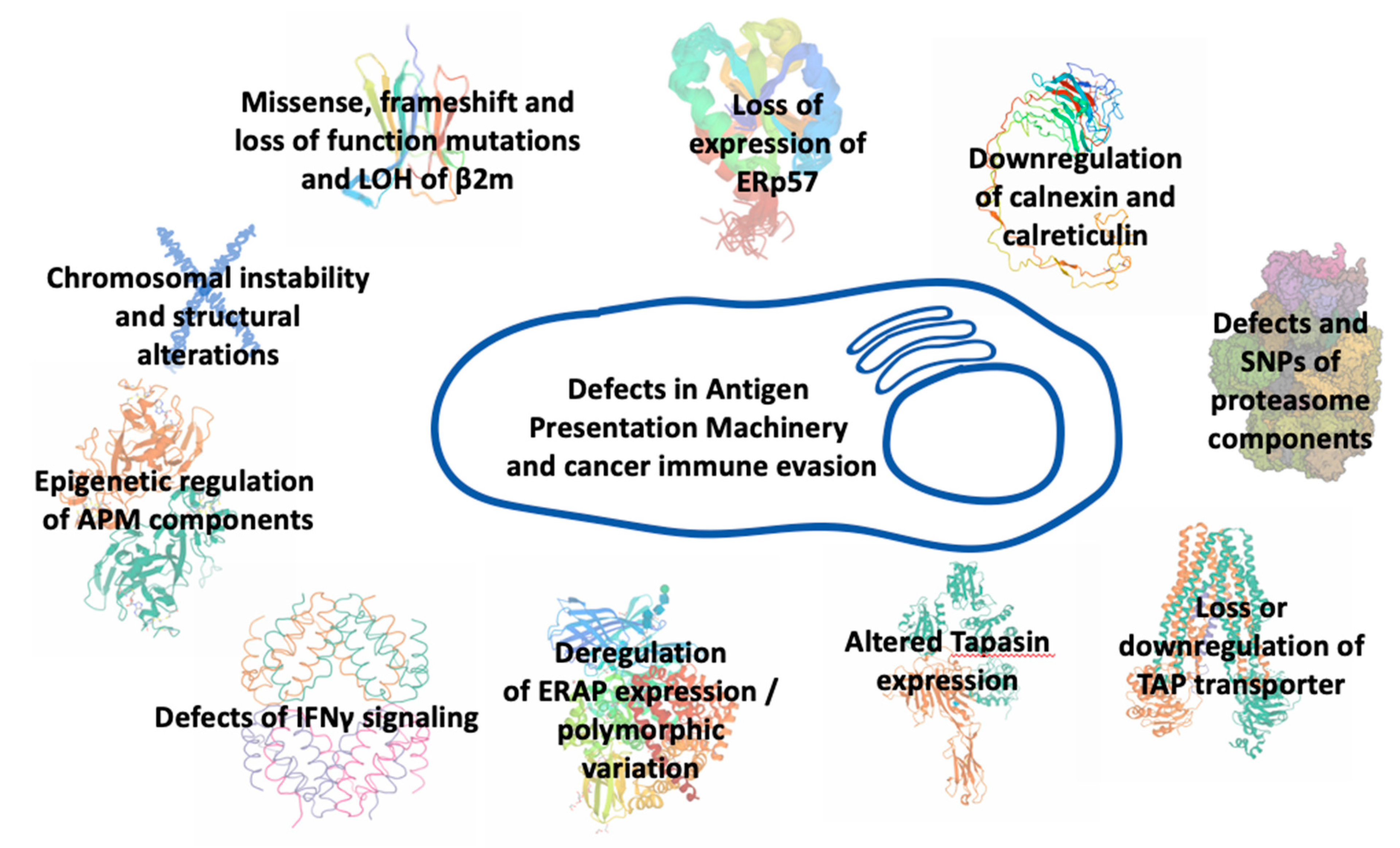
4. Antigen Processing and Presentation in Cancer
5. MHCI Expression in Cancers
6. Dendritic Cells and Cross-Presentation in Cancer
7. The Immunopeptidome and Cancer
8. Tumor Antigens and Tumor-Associated Antigenic Peptides
9. Epigenetic Control of Tumor Antigen Expression and Presentation
10. Neoantigens
11. T Cell Epitopes Associated with Impaired Peptide Processing
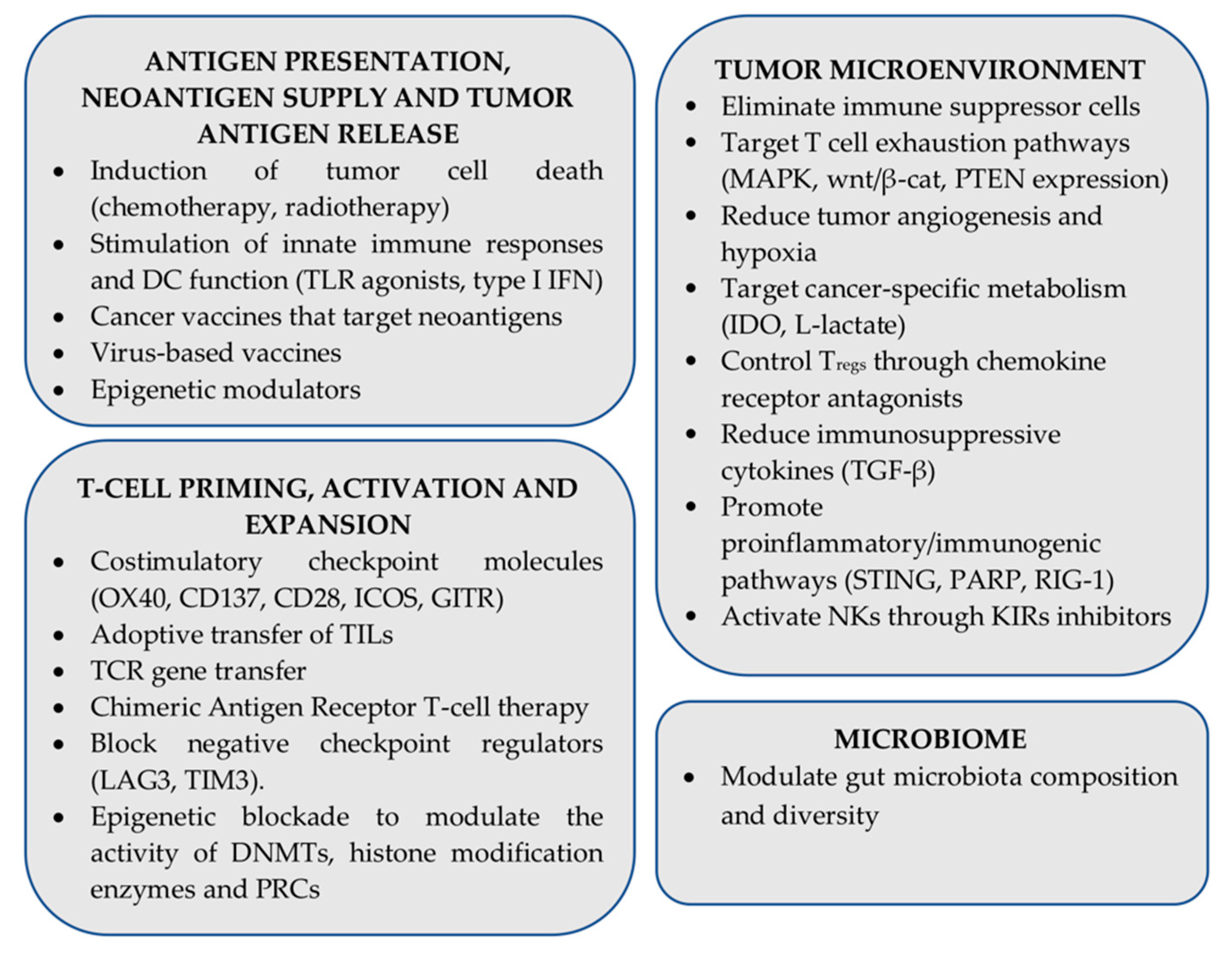
12. Strategies for Enhancing ICI Therapy Effectiveness: The Role of Antigen Presentation
13. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | antigen presenting cell |
| APP(M) | antigen processing and presentation (machinery) |
| CTA | cancer testis antigen |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| DC | dendritic cell |
| HLA | human leukocyte antigen |
| IC(I) | immune checkpoint (inhibitor) |
| LN | lymph node |
| MMR | mismatch repair |
| NK | natural killer cell |
| NSCLC | non-small cell lung cancer |
| PAMPs/DAMPs | pathogen/damage associated molecular patterns |
| PD-1 | programmed cell death protein 1 |
| MHCI | Major Histocompatibility Complex class I |
| TCR | T cell receptor |
| TIL | Tumor infiltrating Lymphocyte |
| TMB | tumor mutational burden |
| TME | tumor microenvironment |
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Gajewski, T.F. Mechanisms of Tumor Cell-Intrinsic Immune Evasion. Annu. Rev. Cancer Biol. 2018, 2, 213–228. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC Regulates the Anti-Tumor Immune Response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Al Bakir, M.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen directed immune escape in lung cancer evolution Rachel. Nature 2020, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Dupage, M.; Mazumbar, C.; Schmidt, L.M.; Cheung, A.F.; Jacks, T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012, 482, 405–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Shea, L.K.; Hundal, J.; et al. Cancer Exome Analysis Reveals a T Cell Dependent Mechanism of Cancer Immunoediting Hirokazu. Nature 2013, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic HLA class I loss is a widespread mechanism of immune evasion which refines the use of tumor mutational burden as a biomarker of checkpoint inhibitor response. Cancer Discov. 2020. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef] [Green Version]
- Vigan, S.; Perreau, M.; Pantaleo, G.; Harari, A. Positive and negative regulation of cellular immune responses in physiologic conditions and diseases. Clin. Dev. Immunol. 2012, 2012. [Google Scholar] [CrossRef]
- Chen, L. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors [published erratum appears in Immunity 1995 Feb;2(2):following 203]. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Van Der Merwe, A.P.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. Affinity and Very Fast Kinetics. J. Exp. Med. 1997, 185, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egen, J.G.; Allison, J.P. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 2002, 16, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.; Gajewski, T.F.; Mackensen, A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: Implications for tumor immunotherapy. Cancer Immunol. Immunother. 2005, 54, 307–314. [Google Scholar] [CrossRef]
- Weiner, L.; Murray, J.; Shuptrine, C. Antibody-based immunotherapy of cancer: New insights, new targets. Cell 2012, 148, 1081–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2016, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvistborg, P.; Philips, D.; Kelderman, S.; Hageman, L.; Ottensmeier, C.; Joseph-Pietras, D.; Welters, M.J.P.; Van Der Burg, S.; Kapiteijn, E.; Michielin, O.; et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci. Transl. Med. 2014, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yost, K.; Satpathy, A.; Wells, D.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.; Granja, J.; Sarin, K.; Brown, R.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Godec, J.; Odorizzi, P.M.; Brown, K.E.; Yates, K.B.; Ngiow, S.F.; Burke, K.P.; Maleri, S.; Grande, S.M.; Francisco, L.M.; et al. The PD-1 Pathway Regulates Development and Function of Memory CD8+ T Cells following Respiratory Viral Infection. Cell Rep. 2020, 31, 107827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ave, H. Advances of FDA A pproved D rugs that T arget PD-1 and PD-L1 for Cancer Immunotherapy. Am. J. Cancer Sci. 2019, 7, 18–31. [Google Scholar]
- Ramagopal, U.A.; Liu, W.; Garrett-Thomson, S.C.; Bonanno, J.B.; Yan, Q.; Srinivasan, M.; Wong, S.C.; Bell, A.; Mankikar, S.; Rangan, V.S.; et al. Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab. Proc. Natl. Acad. Sci. USA 2017, 114, 4223–4232. [Google Scholar] [CrossRef] [Green Version]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, L.; Tsoi, J.; Wang, X.; Emerson, R.; Homet, B.; Chodon, T.; Mok, S.; Huang, R.R.; Cochran, A.J.; Comin-Anduix, B.; et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res. 2014, 20, 2424–2432. [Google Scholar] [CrossRef] [Green Version]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Emma, J.; Taylor, M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; He, Z.; Wang, X.; Li, H.; Liu, X.S. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. Elife 2019, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Blum, S.J. Pathways of Antigen Processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Hollý, J. DRiPs get molecular. Curr. Opin. Immunol. 2020, 64, 130–136. [Google Scholar] [CrossRef]
- Rock, K.L.; York, I.A.; Goldberg, A.L. Post-proteasomal antigen processing for major histocompatibility complex class I presentation. Nat. Immunol. 2004, 5, 670–677. [Google Scholar] [CrossRef]
- Jensen, P.E. Recent advances in antigen processing and presentation. Nat. Immunol. 2007, 8, 1041–1048. [Google Scholar] [CrossRef]
- Blees, A.; Januliene, D.; Hofmann, T.; Koller, N.; Schmidt, C.; Trowitzsch, S.; Moeller, A.; Tampé, R. Structure of the human MHC-I peptide-loading complex. Nature 2017, 551, 525–528. [Google Scholar] [CrossRef]
- Lee, M.Y.; Jeon, J.W.; Sievers, C.; Allen, C.T. Antigen processing and presentation in cancer immunotherapy. J. Immunother. Cancer 2020, 8, 1–15. [Google Scholar] [CrossRef]
- Saveanu, L.; Carroll, O.; Lindo, V.; Del Val, M.; Lopez, D.; Lepelletier, Y.; Greer, F.; Schomburg, L.; Fruci, D.; Niedermann, G.; et al. Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat. Immunol. 2005, 6, 689–697. [Google Scholar] [CrossRef]
- Morozov, G.I.; Zhao, H.; Mage, M.G.; Boyd, L.F.; Jiang, J.; Dolan, M.A.; Venna, R.; Norcross, M.A.; McMurtrey, C.P.; Hildebrand, W.; et al. Interaction of TAPBPR, a tapasin homolog, with MHC-I molecules promotes peptide editing. Proc. Natl. Acad. Sci. USA 2016, 113, E1006–E1015. [Google Scholar] [CrossRef] [Green Version]
- Seliger, B.; Maeurer, M.J.; Ferrone, S. Antigen-processing machinery breakdown and tumor growth. Immunol. Today 2000, 21, 455–464. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wangmo, D.; Robertson, M.; Subramanian, S. Acquired resistance to immune checkpoint blockade therapies. Cancers 2020, 12, 1161. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.; Cho, H.; Perry, C.; Song, J.; Ylaya, K.; Lee, H.; Kim, J.H. Downregulation of ERp57 expression is associated with poor prognosis in early-stage cervical cancer. Biomarkers 2013, 18, 573–579. [Google Scholar] [CrossRef]
- Hasim, A.; Abudula, M.; Aimiduo, R.; Ma, J.Q.; Jiao, Z.; Akula, G.; Wang, T.; Abudula, A. Post-Transcriptional and Epigenetic Regulation of Antigen Processing Machinery (APM) Components and HLA-I in Cervical Cancers from Uighur Women. PLoS ONE 2012, 7, e44952. [Google Scholar] [CrossRef] [Green Version]
- Leys, C.M.; Nomura, S.; LaFleur, B.J.; Ferrone, S.; Kaminishi, M.; Montgomery, E.; Goldenring, J.R. Expression and prognostic significance of prothymosin-α and ERp57 in human gastric cancer. Surgery 2007, 141, 41–50. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Cathro, H.P.; Smolkin, M.E.; Theodorescu, D.; Jo, V.Y.; Ferrone, S.; Frierson, H.F., Jr. Relationship between HLA class I antigen processing machinery component expression and the clinicopathologic characteristics of bladder carcinomas. Cancer Immunol. Immunother. 2012, 59, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Arshad, N.; Cresswell, P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J. Biol. Chem. 2018, 293, 9555–9569. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.M.; Jordanova, E.S.; Kenter, G.G.; Ferrone, S.; Fleuren, G.J. Association of antigen processing machinery and HLA class I defects with clinicopathological outcome in cervical carcinoma. Cancer Immunol. Immunother. 2008, 57, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seliger, B.; Höhne, A.; Knuth, A.; Bernhard, H.; Ehring, B.; Tampé, R.; Huber, C. Reduced membrane major histocompatibility complex class I density and stability in a subset of human renal cell carcinomas with low TAP and LMP expression. Clin. Cancer Res. 1996, 2, 1427–1433. [Google Scholar] [PubMed]
- Ling, A.; Löfgren-Burström, A.; Larsson, P.; Li, X.; Wikberg, M.L.; Öberg, Å.; Stenling, R.; Edin, S.; Palmqvist, R. TAP1 down-regulation elicits immune escape and poor prognosis in colorectal cancer. Oncoimmunology 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, M.; Reichert, T.E.; Kunkel, M.; Gooding, W.; Whiteside, T.L.; Ferrone, S.; Seliger, B. Defects in the human leukocyte antigen class I antigen-processing machinery in head and neck squamous cell carcinoma: Association with clinical outcome. Clin. Cancer Res. 2005, 11, 2552–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaridou, M.-F.; Massa, C.; Handke, D.; Mueller, A.; Friedrich, M.; Subbarayan, K.; Tretbar, S.; Dummer, R.; Koelblinger, P.; Seliger, B. Identification of microRNAs Targeting the Transporter Associated with Antigen Processing TAP1 in Melanoma. J. Clin. Med. 2020, 9, 2690. [Google Scholar] [CrossRef]
- Dissemond, J.; Kothen, T.; Mörs, J.; Weimann, T.K.; Lindeke, A.; Goos, M.; Wagner, S.N. Downregulation of tapasin expression in progressive human malignant melanoma. Arch. Derm. Res. 2003, 295, 43–49. [Google Scholar] [CrossRef]
- Shionoya, Y.; Kanaseki, T.; Miyamoto, S.; Tokita, S.; Hongo, A.; Kikuchi, Y.; Kochin, V.; Watanabe, K.; Horibe, R.; Saijo, H.; et al. Loss of tapasin in human lung and colon cancer cells and escape from tumor-associated antigen-specific CTL recognition. Oncoimmunology 2017, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sokol, L.; Koelzer, V.H.; Rau, T.T.; Karamitopoulou, E.; Zlobec, I.; Lugli, A. Loss of tapasin correlates with diminished CD8+ T-cell immunity and prognosis in colorectal cancer. J. Transl. Med. 2015, 13, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Turnquist, H.R.; Kohlgraf, K.G.; McIlhaney, M.M.; Lee Mosley, R.; Hollingsworth, M.A.; Solheim, J.C. Tapasin decreases immune responsiveness to a model tumor antigen. J. Clin. Immunol. 2004, 24, 462–470. [Google Scholar] [CrossRef]
- Chang, C.C.; Pirozzi, G.; Wen, S.H.; Chung, I.H.; Chiu, B.L.; Errico, S.; Luongo, M.; Lombardi, M.L.; Ferrone, S. Multiple structural and epigenetic defects in the human leukocyte antigen class I antigen presentation pathway in a recurrent metastatic melanoma following immunotherapy. J. Biol. Chem. 2015, 290, 26562–26575. [Google Scholar] [CrossRef] [Green Version]
- Fruci, D.; Ferracuti, S.; Limongi, M.Z.; Cunsolo, V.; Giorda, E.; Fraioli, R.; Sibilio, L.; Carroll, O.; Hattori, A.; van Endert, P.M.; et al. Expression of Endoplasmic Reticulum Aminopeptidases in EBV-B Cell Lines from Healthy Donors and in Leukemia/Lymphoma, Carcinoma, and Melanoma Cell Lines. J. Immunol. 2006, 176, 4869–4879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compagnone, M.; Cifaldi, L.; Fruci, D. Regulation of ERAP1 and ERAP2 genes and their disfunction in human cancer. Hum. Immunol. 2019, 80, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Y.; Yan, Z.; Dai, S.; Liu, S.; Wang, X.; Wang, J.; Zhang, X.; Shi, L.; Yao, Y. Polymorphisms in endoplasmic reticulum aminopeptidase genes are associated with cervical cancer risk in a Chinese Han population. BMC Cancer 2020, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stratikos, E.; Stamogiannos, A.; Zervoudi, E.; Fruci, D. A role for naturally occurring alleles of Endoplasmic Reticulum Aminopeptidases in tumor immunity and cancer predisposition. Front. Oncol. 2014, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- López de Castro, J.A. How ERAP1 and ERAP2 Shape the Peptidomes of Disease-Associated MHC-I Proteins. Front. Immunol. 2018, 9, 2463. [Google Scholar] [CrossRef]
- Koumantou, D.; Barnea, E.; Martin-Esteban, A.; Maben, Z.; Papakyriakou, A.; Mpakali, A.; Kokkala, P.; Pratsinis, H.; Georgiadis, D.; Stern, L.J.; et al. Editing the immunopeptidome of melanoma cells using a potent inhibitor of endoplasmic reticulum aminopeptidase 1 (ERAP1). Cancer Immunol. Immunother. 2019, 68, 1245–1261. [Google Scholar] [CrossRef]
- Mehta, A.M.; Jordanova, E.S.; Corver, W.E.; van Wezel, T.; Uh, H.-W.; Kenter, G.G.; Fleuren, G.J. Single Nucleotide Polymorphisms in Antigen Processing Machinery Component ERAP1 Significantly Associate with Clinical Outcome in Cervical Carcinoma. Genes. Chromosomes Cancer 2009, 48, 410–418. [Google Scholar] [CrossRef]
- Kamphausen, E.; Kellert, C.; Abbas, T.; Akkad, N.; Tenzer, S.; Pawelec, G.; Schild, H.; Van Endert, P.; Seliger, B. Distinct molecular mechanisms leading to deficient expression of ER-resident aminopeptidases in melanoma. Cancer Immunol. Immunother. 2010, 59, 1273–1284. [Google Scholar] [CrossRef]
- Cifaldi, L.; Romania, P.; Lorenzi, S.; Locatelli, F.; Fruci, D. Role of endoplasmic reticulum aminopeptidases in health and disease: From infection to cancer. Int. J. Mol. Sci. 2012, 13, 8338–8352. [Google Scholar] [CrossRef] [Green Version]
- James, E.; Bailey, I.; Sugiyarto, G.; Elliott, T. Induction of Protective Antitumor Immunity through Attenuation of ERAAP Function. J. Immunol. 2013, 190, 5839–5846. [Google Scholar] [CrossRef] [Green Version]
- Zervoudi, E.; Saridakis, E.; Birtley, J.R.; Seregin, S.S.; Reeves, E.; Kokkala, P.; Aldhamen, Y.A.; Amalfitano, A.; Mavridis, I.M.; James, E.; et al. Rationally designed inhibitor targeting antigentrimming aminopeptidases enhances antigen presentation and cytotoxic T-cell responses. Proc. Natl. Acad. Sci. USA 2013, 110, 19890–19895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, M.; Ebstein, F.; Bürger, E.; Textoris-Taube, K.; Gorny, X.; Urban, S.; Zhao, F.; Dannenberg, T.; Sucker, A.; Keller, C.; et al. The proteasome immunosubunits, PA28 and ER-aminopeptidase 1 protect melanoma cells from efficient MART-126-35-specific T-cell recognition. Eur. J. Immunol. 2015, 45, 3257–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifaldi, L.; Romania, P.; Falco, M.; Lorenzi, S.; Meazza, R.; Petrini, S.; Andreani, M.; Pende, D.; Locatelli, F.; Fruci, D. ERAP1 regulates natural killer cell function by controlling the engagement of inhibitory receptors. Cancer Res. 2015, 75, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifaldi, L.; Lo Monaco, E.; Forloni, M.; Giorda, E.; Lorenzi, S.; Petrini, S.; Tremante, E.; Pende, D.; Locatelli, F.; Giacomini, P.; et al. Natural killer cells efficiently reject lymphoma silenced for the endoplasmic reticulum aminopeptidase associated with antigen processing. Cancer Res. 2011, 71, 1597–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, I.C.; Kao, H.K.; Huang, Y.; Wang, C.I.; Yi, J.S.; Liang, Y.; Liao, C.T.; Yen, T.C.; Wu, C.C.; Chang, K.P. Endoplasmic reticulum aminopeptidase 2 involvement in metastasis of oral cavity squamous cell carcinoma discovered by proteome profiling of primary cancer cells. Oncotarget 2017, 8, 61698–61708. [Google Scholar] [CrossRef] [Green Version]
- Warthan, M.D.; Washington, S.L.; Franzese, S.E.; Ramus, R.M.; Kim, K.R.; York, T.P.; Stratikos, E.; Strauss, J.F.; Lee, E.D. The role of endoplasmic reticulum aminopeptidase 2 in modulating immune detection of choriocarcinoma. Biol. Reprod. 2018, 98, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In Vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.W.; Chen-Harris, H.; Mayba, O.; Lianoglou, S.; Wuster, A.; Bhangale, T.; Khan, Z.; Mariathasan, S.; Daemen, A.; Reeder, J.; et al. Germline genetic polymorphisms influence tumor gene expression and immune cell infiltration. Proc. Natl. Acad. Sci. USA 2018, 115, E11701–E11710. [Google Scholar] [CrossRef] [Green Version]
- Reeves, E.; Wood, O.; Ottensmeier, C.H.; King, E.V.; Thomas, G.J.; Elliott, T.; James, E. HPV Epitope Processing Differences Correlate with ERAP1 Allotype and Extent of CD8þ T-cell Tumor Infiltration in OPSCC. Cancer Immunol. Res. 2019, 7, 1202–1213. [Google Scholar] [CrossRef] [Green Version]
- Reeves, E.; Islam, Y.; James, E. ERAP1: A Potential Therapeutic Target for a Myriad of Diseases. Expert Opin. Ther. Targets 2020, 24, 535–544. [Google Scholar] [CrossRef]
- Stratikos, E. Modulating antigen processing for cancer immunotherapy. Oncoimmunology 2014, 3, 2–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2016, 7, 188–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martini, M.; Testi, M.G.; Pasetto, M.; Picchio, M.C.; Innamorati, G.; Mazzocco, M.; Ugel, S.; Cingarlini, S.; Bronte, V.; Zanovello, P.; et al. IFN-γ-mediated upmodulation of MHC class I expression activates tumor-specific immune response in a mouse model of prostate cancer. Vaccine 2010, 28, 3548–3557. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Johannessen, B.; Tengs, T.; Danielsen, S.A.; Eilertsen, I.A.; Lind, G.E.; Berg, K.C.G.; Leithe, E.; Meza-Zepeda, L.A.; Domingo, E.; et al. Multilevel genomics of colorectal cancers with microsatellite instability-clinical impact of JAK1 mutations and consensus molecular subtype 1. Genome Med. 2017, 9, 1–16. [Google Scholar] [CrossRef]
- Karpf, A.R.; Jones, D.A. Reactivating the expression of methylation silenced genes in human cancer. Oncogene 2002, 21, 5496–5503. [Google Scholar] [CrossRef] [Green Version]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.C.; Kersbergen, A.; Lam, E.Y.N.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019, 36, 385–401.e8. [Google Scholar] [CrossRef] [Green Version]
- Campoli, M.; Ferrone, S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008, 27, 5869–5885. [Google Scholar] [CrossRef] [Green Version]
- Setiadi, A.F.; Omilusik, K.; David, M.D.; Seipp, R.P.; Hartikainen, J.; Gopaul, R.; Choi, K.B.; Jefferies, W.A. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008, 68, 9601–9607. [Google Scholar] [CrossRef] [Green Version]
- Zingg, D.; Arenas-Ramirez, N.; Sahin, D.; Rosalia, R.A.; Antunes, A.T.; Haeusel, J.; Sommer, L.; Boyman, O. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017, 20, 854–867. [Google Scholar] [CrossRef]
- Dunn, G.P.; Sheehan, K.C.F.; Old, L.J.; Schreiber, R.D. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res. 2005, 65, 3447–3453. [Google Scholar] [CrossRef] [Green Version]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, C.; Fan, K.; Paschen, A.; Reker Hardrup, S.; Ferrone, S.; Nghiem, P.; Ugurel, S.; Schrama, D.; Becker, J.C. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugurel, S.; Spassova, I.; Wohlfarth, J.; Drusio, C.; Cherouny, A.; Melior, A.; Sucker, A.; Zimmer, L.; Ritter, C.; Schadendorf, D.; et al. MHC class-I downregulation in PD-1/PD-L1 inhibitor refractory Merkel cell carcinoma and its potential reversal by histone deacetylase inhibition: A case series. Cancer Immunol. Immunother. 2019, 68, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Modur, V.; Senovilla, L.; Kroemer, G.; Komurov, K. Suppression of tumor antigen presentation during aneuploid tumor evolution contributes to immune evasion. Oncoimmunology 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Angell, T.E.; Lechner, M.G.; Jang, J.K.; LoPresti, J.S.; Epstein, A.L. MHC class I loss is a frequent mechanism of immune escape in papillary thyroid cancer that is reversed by interferon and selumetinib treatment in Vitro. Clin. Cancer Res. 2014, 20, 6034–6044. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, C.F.W.; Jordanova, E.S.; Zomerdijk-Nooijen, Y.A.; Ter Haar, N.T.; Peters, A.A.W.; Fleuren, G.J. Frequent HLA class I loss is an early event in cervical carcinogenesis. Hum. Immunol. 2005, 66, 1167–1173. [Google Scholar] [CrossRef]
- Ferrone, S.; Marincola, F.M. Loss of HLA class I antigens by melanoma cells: Molecular mechanisms, functional significance and clinical relevance. Trends Neurosci. 1993, 16, 487–494. [Google Scholar] [CrossRef]
- Aptsiauri, N.; Ruiz-Cabello, F.; Garrido, F. The transition from HLA-I positive to HLA-I negative primary tumors: The road to escape from T-cell responses. Curr. Opin. Immunol. 2018, 51, 123–132. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N. Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology 2019, 158, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Garrido, F.; Perea, F.; Bernal, M.; Sánchez-Palencia, A.; Aptsiauri, N.; Ruiz-Cabello, F. The escape of cancer from T cell-mediated immune surveillance: HLA class I loss and tumor tissue architecture. Vaccines 2017, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochan, G.; Escors, D.; Breckpot, K.; Guerrero-Setas, D. Role of non-classical MHC class I molecules in cancer immunosuppression. Oncoimmunology 2013, 2, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, J.A. HLA-mediated tumor escape mechanisms that may impair immunotherapy clinical outcomes via T-cell activation (Review). Oncol. Lett. 2017, 14, 4415–4427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, S.D.; Chen, Z.; Melendez, B.; Talukder, A.; Khalili, J.S.; Rodriguez-Cruz, T.; Liu, S.; Whittington, M.; Deng, W.; Li, F.; et al. BRAFV600E co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol. Res. 2015, 3, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Cui, X.; Zhuo, Y.; Li, H.; Ha, C.; Xin, L.; Ren, Y.; Zhang, W.; Sun, X.; et al. Oncoprotein SND1 hijacks nascent MHC-I heavy chain to ER-associated degradation, leading to impaired CD8+ T cell response in tumor. Sci. Adv. 2020, 6, 1–16. [Google Scholar] [CrossRef]
- Romero, I.; Martinez, M.; Garrido, C.; Collado, A.; Algarra, I.; Garrido, F.; Garcia-Lora, A.M. The tumour suppressor Fhit positively regulates MHC class I expression on cancer cells. J. Pathol. 2012, 227, 367–379. [Google Scholar] [CrossRef]
- Garancher, A.; Suzuki, H.; Haricharan, S.; Chau, L.Q.; Masihi, M.B.; Rusert, J.M.; Norris, P.S.; Carrette, F.; Romero, M.M.; Morrissy, S.A.; et al. Tumor necrosis factor overcomes immune evasion in p53-mutant medulloblastoma. Nat. Neurosci. 2020, 1–12. [Google Scholar] [CrossRef]
- Kriegsman, B.A.; Vangala, P.; Chen, B.J.; Meraner, P.; Brass, A.L.; Garber, M.; Rock, K.L. Frequent Loss of IRF2 in Cancers Leads to Immune Evasion through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. J. Immunol. 2019, 203, 1999–2010. [Google Scholar] [CrossRef]
- Huang, L.; Malu, S.; McKenzie, J.A.; Andrews, M.C.; Talukder, A.H.; Tieu, T.; Karpinets, T.; Haymaker, C.; Forget, M.A.; Williams, L.J.; et al. The RNA-binding protein MEX£B mediates resistance to cancer immunotherapy by downregulating HLA-a expression. Clin. Cancer Res. 2018, 24, 3366–3376. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xiong, H.G.; Xiao, Y.; Yang, Q.C.; Yang, S.C.; Tang, H.C.; Zhang, W.F.; Sun, Z.J. Long Non-coding RNA LINC02195 as a Regulator of MHC I Molecules and Favorable Prognostic Marker for Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kulski, J.K. Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations. Cells 2019, 8, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Lin, T.Y.; Chen, L.; Liu, Y.; Dian, M.J.; Hao, W.C.; Lin, X.L.; Li, X.Y.; Li, Y.L.; Lian, M.; et al. Mir-19 regulates the expression of interferon-induced genes and mhc class i genes in human cancer cells. Int. J. Med. Sci. 2020, 17, 953–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichmüller, S.B.; Osen, W.; Mandelboim, O.; Seliger, B. Immune Modulatory microRNAs Involved in Tumor Attack and Tumor Immune Escape. J. Natl. Cancer Inst. 2017, 109, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Samstein, R.M.; Valero, C.; Chan, T.A.; Morris, L.G.T. Tumor mutational burden as a predictive biomarker for checkpoint inhibitor immunotherapy. Hum. Vaccines Immunother. 2020, 16, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Stokidis, S.; Fortis, S.P.; Kogionou, P.; Anagnostou, T.; Perez, S.A.; Baxevanis, C.N. HLA class I allele expression and clinical outcome in De Novo metastatic prostate cancer. Cancers 2020, 12, 1623. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2017, 47, 549–562. [Google Scholar] [CrossRef] [Green Version]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell 2017, 171, 1272–1283.e15. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, A.A.; Kwon, E.D.; Van Elsas, A. Costimulatory wars: The tumor menace. Curr. Opin. Immunol. 2000, 12, 589–596. [Google Scholar] [CrossRef]
- Kurts, C.; Cannarile, M.; Klebba, I.; Brocker, T. Cutting Edge: Dendritic Cells Are Sufficient to Cross-Present Self-Antigens to CD8 T Cells In Vivo. J. Immunol. 2001, 166, 1439–1442. [Google Scholar] [CrossRef] [Green Version]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.B.; Schuler, G. Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance or immunity? Trends Immunol. 2002, 23, 445–449. [Google Scholar] [CrossRef]
- Reis, E.; Sousa, C. Dendritic cells in a mature age. Nat. Rev. Immunol. 2006, 6, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Worbs, T.; Hammerschmidt, S.I.; Förster, R. Dendritic cell migration in health and disease. Nat. Rev. Immunol. 2017, 17, 30–48. [Google Scholar] [CrossRef]
- Gardner Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [CrossRef] [Green Version]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Ma, Y.; Shurin, G.V.; Peiyuan, Z.; Shurin, M.R. Dendritic cells in the cancer microenvironment. J. Cancer 2013, 4, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.N.; Gebhardt, T.; Carbone, F.R.; Heath, W.R. Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol. 2013, 31, 137–161. [Google Scholar] [CrossRef]
- Preynat-Seauve, O.; Schuler, P.; Contassot, E.; Beermann, F.; Huard, B.; French, L.E. Tumor-Infiltrating Dendritic Cells Are Potent Antigen-Presenting Cells Able to Activate T Cells and Mediate Tumor Rejection. J. Immunol. 2006, 176, 61–67. [Google Scholar] [CrossRef]
- van Mierlo, G.J.D.; Boonman, Z.F.H.M.; Dumortier, H.M.H.; den Boer, A.T.; Fransen, M.F.; Nouta, J.; van der Voort, E.I.H.; Offringa, R.; Toes, R.E.M.; Melief, C.J.M. Activation of Dendritic Cells That Cross-Present Tumor-Derived Antigen Licenses CD8 + CTL to Cause Tumor Eradication. J. Immunol. 2004, 173, 6753–6759. [Google Scholar] [CrossRef] [Green Version]
- Henrickson, S.E.; Perro, M.; Loughhead, S.M.; Senman, B.; Quigley, M.; Alexe, G.; Iannacone, M.; Flynn, M.P.; Jesneck, J.L.; Imam, S.; et al. Antigen availability determines CD8+ T cell-dendritic cell interaction kinetics and memory fate decisions. Immunity 2014, 39, 496–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaccorsi, I.; Campana, S.; Morandi, B.; Ferlazzo, G. Acquisition and Presentation of Tumor Antigens by Dendritic Cells. Crit. Rev. Immunol. 2015, 35, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Sofopoulos, M.; Fortis, S.P.; Vaxevanis, C.K.; Sotiriadou, N.N.; Arnogiannaki, N.; Ardavanis, A.; Vlachodimitropoulos, D.; Perez, S.A.; Baxevanis, C.N. The prognostic significance of peritumoral tertiary lymphoid structures in breast cancer. Cancer Immunol. Immunother. 2019, 68, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T. Getting to the source: Dendritic cells as therapeutic reagents for the treatment of patients with cancer. Ann. Surg. 1997, 226, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Stoitzner, P.; Green, L.K.; Jung, J.Y.; Price, K.M.; Atarea, H.; Kivell, B.; Ronchese, F. Inefficient presentation of tumor-derived antigen by tumor-infiltrating dendritic cells. Cancer Immunol. Immunother. 2008, 57, 1665–1673. [Google Scholar] [CrossRef]
- Mcdonnell, A.M.; Lesterhuis, W.J.; Khong, A.; Nowak, A.K.; Lake, R.A.; Currie, A.J.; Robinson, B.W.S. Tumor-infiltrating dendritic cells exhibit defective cross-presentation of tumor antigens, but is reversed by chemotherapy. Eur. J. Immunol. 2015, 45, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Zhuang, Y.; Liu, C.; Liu, J.; Li, G. Resistance mechanism of PD-1/PD-L1 blockade in the cancer-immunity cycle. Onco. Targets 2020, 13, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Scarlett, U.K.; Rutkowski, M.R.; Rauwerdink, A.M.; Fields, J.; Escovar-Fadul, X.; Baird, J.; Cubillos-Ruiz, J.R.; Jacobs, A.C.; Gonzalez, J.L.; Weaver, J.; et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 2012, 209, 495–506. [Google Scholar] [CrossRef]
- Sánchez-Paulete, A.R.; Teijeira, A.; Cueto, F.J.; Garasa, S.; Pérez-Gracia, J.L.; Sánchez-Arráez, A.; Sancho, D.; Melero, I. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann. Oncol. 2017, 28, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Bandola-Simon, J.; Roche, P.A. Dysfunction of antigen processing and presentation by dendritic cells in cancer. Mol. Immunol. 2019, 113, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Baxevanis, C.N.; Fortis, S.P.; Perez, S.A. The balance between breast cancer and the immune system: Challenges for prognosis and clinical benefit from immunotherapies. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Cao, W.; Ramakrishnan, R.; Tuyrin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J.; et al. Correction: Oxidized Lipids Block Antigen Cross-Presentation by Dendritic Cells in Cancer. J. Immunol. 2014, 192, 4935. [Google Scholar] [CrossRef] [Green Version]
- Hargadon, K.M. Tumor-altered dendritic cell function: Implications for anti-tumor immunity. Front. Immunol. 2013, 4, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, B.Y.; Bolton, H.; Kim, J.W.; Silveira, P.A.; Fromm, P.D.; Clark, G.J. On the other side: Manipulating the immune checkpoint landscape of dendritic cells to enhance cancer immunotherapy. Front. Oncol. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Alloatti, A.; Rookhuizen, D.C.; Joannas, L.; Carpier, J.M.; Iborra, S.; Magalhaes, J.G.; Yatim, N.; Kozik, P.; Sancho, D.; Albert, M.L.; et al. Critical role for Sec22b-dependent antigen crosspresentation in antitumor immunity. J. Exp. Med. 2017, 214, 2231–2241. [Google Scholar] [CrossRef]
- Sánchez-Paulete, A.R.; Cueto, F.J.; Martínez-lópez, M.; Labiano, S.; Rodridriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, A.M.; Quetglas, J.I.; Sancho, D.; et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires Batf3-dependent dendritic cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Dammeijer, F.; Lau, S.P.; van Eijck, C.H.J.; van der Burg, S.H.; Aerts, J.G.J.V. Rationally combining immunotherapies to improve efficacy of immune checkpoint blockade in solid tumors. Cytokine Growth Factor Rev. 2017, 36, 5–15. [Google Scholar] [CrossRef]
- Kadam, P.; Sharma, S. Pd-1 immune checkpoint blockade promotes therapeutic cancer vaccine to eradicate lung cancer. Vaccines 2020, 8, 317. [Google Scholar] [CrossRef] [PubMed]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Orpilla, J.; Moughon, D.; Shin, N.; Sedighim, S.; Yong, W.H.; Li, G.; Cloughesy, T.F.; et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2019, 1, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roenblatt, J.; Glotzbecker, B.; Millis, H.; Baldev, V.; Tzachanis, D.; Levine, J.D.; Joyce, R.M.; Wellenstein, K.; Keefe, W.; Schickler, M.; et al. PD-1 blockade by CT-011, anti PD-1 antibody, enhances ex-vivo T cell responses to autologous dendritic/myeloma fusion vaccine. J. Immunother. 2011, 34, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Xi, H.; Ju, S.; Zhang, X. Blockade of PD-1/PD-L1 immune checkpoint during DC vaccination induces potent protective immunity against breast cancer in hu-SCID mice. Cancer Lett. 2013, 336, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Shimizu, K.; Iyoda, T.; Ueda, S.; Shinga, J.; Mochizuki, Y.; Watanabe, T.; Ohara, O.; Fujii, S. ichiro PD-L1 Expression Affects Neoantigen Presentation. iScience 2020, 23, 101238. [Google Scholar] [CrossRef]
- Linette, G.P.; Becker-Hapak, M.; Skidmore, Z.L.; Baroja, M.L.; Xu, C.; Hundal, J.; Spencer, D.H.; Fu, W.; Cummins, C.; Robnett, M.; et al. Immunological ignorance is an enabling feature of the oligo-clonal T cell response to melanoma neoantigens. Proc. Natl. Acad. Sci. USA 2019, 116, 23662–23670. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Jordan, S.; Casanova-acebes, M.; Agudo, J.; Tung, N.; Chakarov, S.; Hogstad, B.; Bosenberg, M.; et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Kubiniok, P.; Kovalchik, K.; Ma, Q.; Duquette, J.D.; Mongrain, I.; Deutsch, E.W.; Peters, B.; Sette, A.; Sirois, I.; et al. The Human Immunopeptidome Project: A roadmap to predict and treat immune diseases Running title: Next-generation immunopeptidomics. Mol. Cell Proteom. 2020, 19, 31–49. [Google Scholar] [CrossRef]
- Backert, L.; Kowalewski, D.J.; Walz, S.; Schuster, H.; Berlin, C.; Neidert, M.C.; Schemionek, M.; Brümmendorf, T.H.; Vucinic, V.; Niederwieser, D.; et al. A meta-analysis of HLA peptidome composition in different hematological entities: Entity-specific dividing lines and “panleukemia” antigens. Oncotarget 2017, 8, 43915–43924. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Coukos, G. Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr. Opin. Immunol. 2016, 41, 9–17. [Google Scholar] [CrossRef]
- Schuster, H.; Peper, J.K.; Bösmüller, H.C.; Röhle, K.; Backert, L.; Bilich, T.; Ney, B.; Löffler, M.W.; Kowalewski, D.J.; Trautwein, N.; et al. The immunopeptidomic landscape of ovarian carcinomas. Proc. Natl. Acad. Sci. USA 2017, 114, E9942–E9951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffler, M.W.; Kowalewski, D.J.; Backert, L.; Bernhardt, J.; Adam, P.; Schuster, H.; Dengler, F.; Backes, D.; Kopp, H.G.; Beckert, S.; et al. Mapping the HLA ligandome of colorectal cancer reveals an imprint of malignant cell transformation. Cancer Res. 2018, 78, 4627–4641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feola, S.; Chiaro, J.; Martins, B.; Cerullo, V. Uncovering the tumor antigen landscape: What to know about the discovery process. Cancers 2020, 12, 1660. [Google Scholar] [CrossRef]
- Kanaseki, T.; Torigoe, T. Proteogenomics: Advances in cancer antigen research. Immunol. Med. 2019, 42, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Admon, A.; Bassani-Sternberg, M. The human immunopeptidome project, a suggestion for yet another postgenome next big thing. Mol. Cell. Proteom. 2011, 10, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Pfammatter, S.; Bonneil, E.; Lanoix, J.; Vincent, K.; Hardy, M.P.; Courcelles, M.; Perreault, C.; Thibault, P. Extending the Comprehensiveness of Immunopeptidome Analyses Using Isobaric Peptide Labeling. Anal. Chem. 2020, 92, 9194–9204. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van Den Eynde, B.J.; Van Der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Ilyas, S.; Yang, J.C. Landscape of Tumor Antigens in T Cell Immunotherapy. J. Immunol. 2015, 195, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Fritsche, J.; Roszik, J.; Williams, L.J.; Peng, X.; Chiu, Y.; Tsou, C.C.; Hoffgaard, F.; Goldfinger, V.; Schoor, O.; et al. RNA editing derived epitopes function as cancer antigens to elicit immune responses. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Lill, J.R. MHC class I presented antigens from malignancies: A perspective on analytical characterization & immunogenicity. J. Proteom. 2018, 191, 48–57. [Google Scholar] [CrossRef]
- Engelhard, V.H.; Obeng, R.C.; Cummings, K.L.; Petroni, G.R.; Ambakhutwala, A.L.; Chianese-Bullock, K.A.; Smith, K.T.; Lulu, A.; Varhegyi, N.; Smolkin, M.E.; et al. MHC-restricted phosphopeptide antigens: Preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.I.; Yewdell, J.W.; Yang, J.C. Immune recognition of a human renal cancer antigen through post-translational protein splicing. Nature 2004, 427, 252–256. [Google Scholar] [CrossRef]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.H.; Heck, A.J.R.; Mishto, M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016, 354, 354–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mylonas, R.; Beer, I.; Iseli, C.; Chong, C.; Pak, H.S.; Gfeller, D.; Coukos, G.; Xenarios, I.; Müller, M.; Bassani-Sternberg, M. Estimating the contribution of proteasomal spliced peptides to the HLA-I ligandome. Mol. Cell. Proteom. 2018, 17, 2347–2357. [Google Scholar] [CrossRef] [Green Version]
- Chong, C.; Müller, M.; Pak, H.S.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Laumont, C.M.; Daouda, T.; Laverdure, J.P.; Bonneil, É.; Caron-Lizotte, O.; Hardy, M.P.; Granados, D.P.; Durette, C.; Lemieux, S.; Thibault, P.; et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Fratta, E.; Coral, S.; Covre, A.; Parisi, G.; Colizzi, F.; Danielli, R.; Marie Nicolay, H.J.; Sigalotti, L.; Maio, M. The biology of cancer testis antigens: Putative function, regulation and therapeutic potential. Mol. Oncol. 2011, 5, 164–182. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, Z.A.; Whitehurst, A.W. Emerging Contributions of Cancer/Testis Antigens to Neoplastic Behaviors. Trends Cancer 2018, 4, 701–712. [Google Scholar] [CrossRef]
- Faramarzi, S.; Ghafouri-Fard, S. Melanoma: A prototype of cancer-testis antigen-expressing malignancies. Immunotherapy 2017, 9, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Arenas-Ramirez, N.; Sahin, D.; Boyman, O. Epigenetic mechanisms of tumor resistance to immunotherapy. Cell. Mol. Life Sci. 2018, 75, 4163–4176. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Gnjatic, S.; Li, H.; Powel, S.; Gallardo, H.F.; Ritter, E.; Ku, G.Y.; Jungbluth, A.A.; Segal, N.H.; Rasalan, T.S.; et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc. Natl. Acad. Sci. USA 2008, 105, 20410–20415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roszik, J.; Wang, W.-L.; Livingston, J.A.; Roland, C.L.; Ravi, V.; Yee, C.; Hwu, P.; Futreal, A.; Lazar, A.J.; Patel, S.R.; et al. Overexpressed PRAME is a potential immunotherapy target in sarcoma subtypes. Clin. Sarcoma Res. 2017, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishak, C.A.; Classon, M.; De Carvalho, D.D. Deregulation of Retroelements as an Emerging Therapeutic Opportunity in Cancer. Trends Cancer 2018, 4, 583–597. [Google Scholar] [CrossRef]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Wang, X.; Kageshita, T.; Wakasugi, S.; Karpf, A.R.; Ferrone, S. Regulation of high molecular weight-melanoma associated antigen (HMW-MAA) gene expression by promoter DNA methylation in human melanoma cells. Oncogene 2006, 25, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Qamra, A.; Xing, M.; Padmanabhan, N.; Kwok, J.J.T.; Zhang, S.; Xu, C.; Leong, Y.S.; Lim, A.P.L.; Tang, Q.; Ooi, W.F.; et al. Epigenomic promoter alterations amplify gene isoform and immunogenic diversity in gastric adenocarcinoma. Cancer Discov. 2017, 7, 630–651. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, P.K.; Duan, F. Harnessing the antigenic fingerprint of each individual cancer for immunotherapy of human cancer: Genomics shows a new way and its challenges. Cancer Immunol. Immunother. 2013, 62, 967–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vormehr, M.; Diken, M.; Boegel, S.; Kreiter, S.; Türeci, Ö.; Sahin, U. Mutanome directed cancer immunotherapy. Curr. Opin. Immunol. 2016, 39, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yu, L.; Wei, X.; Wei, Y. Role of tumor gene mutations in treatment response to immune checkpoint blockades. Precis. Clin. Med. 2019, 2, 100–109. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Geukes Foppen, M.H.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2016, 352, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, M.; Miller, V.; Stephens, P.J.; Daniels, G.A. Response to Immunotherapy in Diverse Cancers. Mol. Cancer 2018, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subudhi, S.K.; Vence, L.; Zhao, H.; Blando, J.; Yadav, S.S.; Xiong, Q.; Reuben, A.; Aparicio, A.; Corn, P.G.; Chapin, B.F.; et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci. Transl. Med. 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Xu, H.; Xu, X.; Guo, T.; Ge, W. High tumor mutation burden predicts better efficacy of immunotherapy: A pooled analysis of 103078 cancer patients. Oncoimmunology 2019, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Zhang, T.; Li, J.; Lin, J.; Liang, W.; Huang, W.; Wan, N.; Jiang, J. Association between tumor mutation burden (TMB) and outcomes of cancer patients treated with PD-1/ PD-L1 inhibitions: A meta-analysis. Front. Pharm. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowanetz, M.; Zou, W.; Shames, D.S.; Cummings, C.; Rizvi, N.; Spira, A.I.; Frampton, G.M.; Leveque, V.; Flynn, S.; Mocci, S.; et al. Tumor mutation load assessed by FoundationOne (FM1) is associated with improved efficacy of atezolizumab (atezo) in patients with advanced NSCLC. Ann. Oncol. 2016, 27, vi23. [Google Scholar] [CrossRef]
- Van Rooij, N.; Van Buuren, M.M.; Philips, D.; Velds, A.; Toebes, M.; Heemskerk, B.; Van Dijk, L.J.A.; Behjati, S.; Hilkmann, H.; El Atmioui, D.; et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 2013, 31. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e4. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson III, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Alborelli, I.; Leonards, K.; Rothschild, S.I.; Leuenberger, L.P.; Savic Prince, S.; Mertz, K.D.; Poechtrager, S.; Buess, M.; Zippelius, A.; Läubli, H.; et al. Tumor mutational burden assessed by targeted NGS predicts clinical benefit from immune checkpoint inhibitors in non-small cell lung cancer. J. Pathol. 2020, 250, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Xu, J.; Du, C.; Wu, Y.; Xia, D.; Lv, W.; Hu, J. The Predictive Value of Tumor Mutation Burden on Efficacy of Immune Checkpoint Inhibitors in Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, J.E.; Hoff, J.; Powles, T.; Van Der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; Fleming, M.T.; Petrylak, D.P.; Perez-gracia, J.L.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016. [Google Scholar] [CrossRef] [Green Version]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; Van Den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2017, 4, 959–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Gregory, P. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas Theo. Cell Rep. 2018, 23, 239–254. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.K.; Anker, J.F.; Oh, M.S.; Bais, P.; Namburi, S.; Agte, S.; Giles, F.J.; Chuang, J.H. Mutations in DNA repair genes are associated with increased neoantigen burden and a distinct immunophenotype in lung squamous cell carcinoma. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rospo, G.; Lorenzato, A.; Amirouchene-Angelozzi, N.; Magrì, A.; Cancelliere, C.; Corti, G.; Negrino, C.; Amodio, V.; Montone, M.; Bartolini, A.; et al. Evolving neoantigen profiles in colorectal cancers with DNA repair defects. Genome Med. 2019, 11, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magri, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Goldberg, R.M. Hypermutated Tumors and Immune Checkpoint Inhibition. Drugs 2018, 78, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Azad, N.S.; Laheru, D.; Biedrzycki, B.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Marijt, K.A.; Van Der Burg, S.H.; van Hall, T. TEIPP peptides: Exploration of unTAPped cancer antigens. Oncoimmunology 2019, 8, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Marijt, K.A.; Doorduijn, E.M.; van Hall, T. TEIPP antigens for T-cell based immunotherapy of immune-edited HLA class Ilow cancers. Mol. Immunol. 2019, 113, 43–49. [Google Scholar] [CrossRef]
- Seidel, U.J.E.; Oliveira, C.C.; Lampen, M.H.; Van Hall, T. A novel category of antigens enabling CTL immunity to tumor escape variants: Cinderella antigens. Cancer Immunol. Immunother. 2012, 61, 119–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durgeau, A.; El Hage, F.; Vergnon, I.; Validire, P.; de Montpréville, V.; Besse, B.; Soria, J.-C.; van Hall, T.; Mami-Chouaib, F. Different Expression Levels of the TAP Peptide Transporter Lead to Recognition of Different Antigenic Peptides by Tumor-Specific CTL. J. Immunol. 2011, 187, 5532–5539. [Google Scholar] [CrossRef] [Green Version]
- Marijt, K.A.; Blijleven, L.; Verdegaal, E.M.E.; Kester, M.G.; Kowalewski, D.J.; Rammensee, H.G.; Stevanović, S.; Heemskerk, M.H.M.; Van Der Burg, S.H.; Van Hall, T. Identification of non-mutated neoantigens presented by TAP-deficient tumors. J. Exp. Med. 2018, 215, 2325–2337. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, N.A.; Gonzalez, F.; Shastri, N. Non-classical MHC class Ib-restricted cytotoxic T cells monitor antigen processing in the endoplasmic reticulum Niranjana. Nat. Immunol. 2012, 13, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, N.A.; de Verteuil, D.A.; Sriranganadane, D.; Yahyaoui, W.; Thibault, P.; Perreault, C. Shastr ERAAP shapes the peptidome associated with classical and non-classical MHC class I molecules. J. Immunol. 2016, 197, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, N.A.; Shastri, N. Immune surveillance for ERAAP dysfunction. Mol. Immunol. 2013, 55, 120–122. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef]
- Puccini, A.; Battaglin, F.; Iaia, M.L.; Lenz, H.J.; Salem, M.E. Overcoming resistance to anti-PD1 and anti-PD-L1 treatment in gastrointestinal malignancies. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Gao, L.; Liu, Y.; Meng, F.; Li, X.; Qin, F.X.F. Targeting the tumor microenvironment to overcome immune checkpoint blockade therapy resistance. Immunol. Lett. 2020, 220, 88–96. [Google Scholar] [CrossRef]
- Trojaniello, C.; Vitale, M.G.; Scarpato, L.; Esposito, A.; Ascierto, P.A. Melanoma immunotherapy: Strategies to overcome pharmacological resistance. Expert. Rev. Anticancer. 2020, 20, 289–304. [Google Scholar] [CrossRef]
- Huang, J.; Jiang, Z.; Wang, Y.; Fan, X.; Cai, J.; Yao, X.; Liu, L.; Huang, J.; He, J.; Xie, C.; et al. Modulation of gut microbiota to overcome resistance to immune checkpoint blockade in cancer immunotherapy. Curr. Opin. Pharm. 2020, 54, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, A.M.; Stopfer, L.; Lee, S.; Gaglia, G.; Sandel, D.; Santagata, S.; Lin, N.U.; Trepel, J.B.; White, F.; Jacks, T.; et al. Rebalancing protein homeostasis enhances tumor antigen presentation. Clin. Cancer Res. 2019, 25, 6392–6405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tudor Ilca, F.; Neerincx, A.; Wills, M.R.; De La Roche, M.; Boyle, L.H. Utilizing TAPBPR to promote exogenous peptide loading onto cell surface MHC I molecules. Proc. Natl. Acad. Sci. USA 2018, 115, 9353–9361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evnouchidou, I.; Weimershaus, M.; Saveanu, L.; van Endert, P. ERAP1–ERAP2 Dimerization Increases Peptide-Trimming Efficiency. J. Immunol. 2014, 193, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Fruci, D.; Giacomini, P.; Nicotra, M.R.; Forloni, M.; Fraioli, R.; Saveanu, L.; Endert, P.V.A.N. Altered Expression of Endoplasmic Reticulum Aminopeptidases ERAP1 and ERAP2 in Transformed Non-Lymphoid Human Tissues. J. Cell. Physiol. 2008, 216, 742–749. [Google Scholar] [CrossRef]
- Saveanu, L. IRAP Identifies an Endosomal Compartment Required for MHC Class I Cross-Presentation. Science 2009, 325, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Papakyriakou, A.; Zervoudi, E.; Tsoukalidou, S.; Mauvais, F.X.; Sfyroera, G.; Mastellos, D.C.; Van Endert, P.; Theodorakis, E.A.; Vourloumis, D.; Stratikos, E. 3,4-diaminobenzoic acid derivatives as inhibitors of the oxytocinase subfamily of m1 aminopeptidases with immune-regulating properties. J. Med. Chem. 2015, 58, 1524–1543. [Google Scholar] [CrossRef] [Green Version]
- Georgiadis, D.; Ziotopoulou, A.; Kaloumenou, E.; Lelis, A.; Papasava, A. The Discovery of Insulin-Regulated Aminopeptidase (IRAP) Inhibitors: A Literature Review. Front. Pharm. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Liddle, J.; Hutchinson, J.P.; Kitchen, S.; Rowland, P.; Neu, M.; Cecconie, T.; Holmes, D.S.; Jones, E.; Korczynska, J.; Koumantou, D.; et al. Targeting the Regulatory Site of ER Aminopeptidase 1 Leads to the Discovery of a Natural Product Modulator of Antigen Presentation. ACS Appl. Mater. Interfaces 2020. [Google Scholar] [CrossRef]
- Georgiadis, D.; Mpakali, A.; Koumantou, D.; Stratikos, E. Inhibitors of ER Aminopeptidase 1 and 2: From Design to Clinical Application. Curr. Med. Chem. 2019, 26, 1–14. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Company | Target | Indications * |
|---|---|---|---|
| Ipilimumab (Yervoy®) | Bristol-Myers Squibb, New York, U.S.A. | CTLA-4 | Melanoma, RCC, colorectal cancer, HCC, NSCLC, malignant pleural mesothelioma |
| Pembrolizumab (Keytruda®) | Merck Co., New Jersey, U.S.A. | PD-1 | Melanoma, NSCLC, SCLC, HNSCC, urothelial carcinoma, primary mediastinal large B-cell lymphoma, gastric cancer, cervical cancer, esophageal cancer, TNBC, hepatocellular carcinoma, MCC, RCC, endometrial carcinoma, cutaneous squamous cell carcinoma, tumor mutational burden-High cancer, microsatellite instability or mismatch repair deficient colorectal cancer, microsatellite instability-High cancer |
| Nivolumab (Opdivo®) | Bristol-Myers Squibb, New York, U.S.A. | PD-1 | Melanoma, RCC, NSCLC, SCLC, cHL, HNSCC, HCC, colorectal cancer, urothelial carcinoma, esophageal squamous cell carcinoma |
| Cemiplimab (Libtayo®) | Sanofi, Paris, France | PD-1 | Cutaneous squamous cell carcinoma |
| Atezolizumab (Tecentriq®) | Roche, Basel, Switzerland | PD-L1 | Urothelial carcinoma, NSCLC, TNBC, SCLC, HCC, melanoma |
| Avelumab (Bavencio®) | Pfizer, New York, U.S.A. and Merck, U.S.A. | PD-L1 | Metastatic MCC, metastatic urothelial carcinoma |
| Durvalumab (Imfinzi®) | AstraZeneca, Cambridge, U.K. | PD-L1 | Advanced or metastatic urothelial carcinoma, stage III NSCLC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mpakali, A.; Stratikos, E. The Role of Antigen Processing and Presentation in Cancer and the Efficacy of Immune Checkpoint Inhibitor Immunotherapy. Cancers 2021, 13, 134. https://doi.org/10.3390/cancers13010134
Mpakali A, Stratikos E. The Role of Antigen Processing and Presentation in Cancer and the Efficacy of Immune Checkpoint Inhibitor Immunotherapy. Cancers. 2021; 13(1):134. https://doi.org/10.3390/cancers13010134
Chicago/Turabian StyleMpakali, Anastasia, and Efstratios Stratikos. 2021. "The Role of Antigen Processing and Presentation in Cancer and the Efficacy of Immune Checkpoint Inhibitor Immunotherapy" Cancers 13, no. 1: 134. https://doi.org/10.3390/cancers13010134