Evolution of the Experimental Models of Cholangiocarcinoma

 ,
,
Abstract
:1. Introduction
2. Evolution of in vitro CCA Models
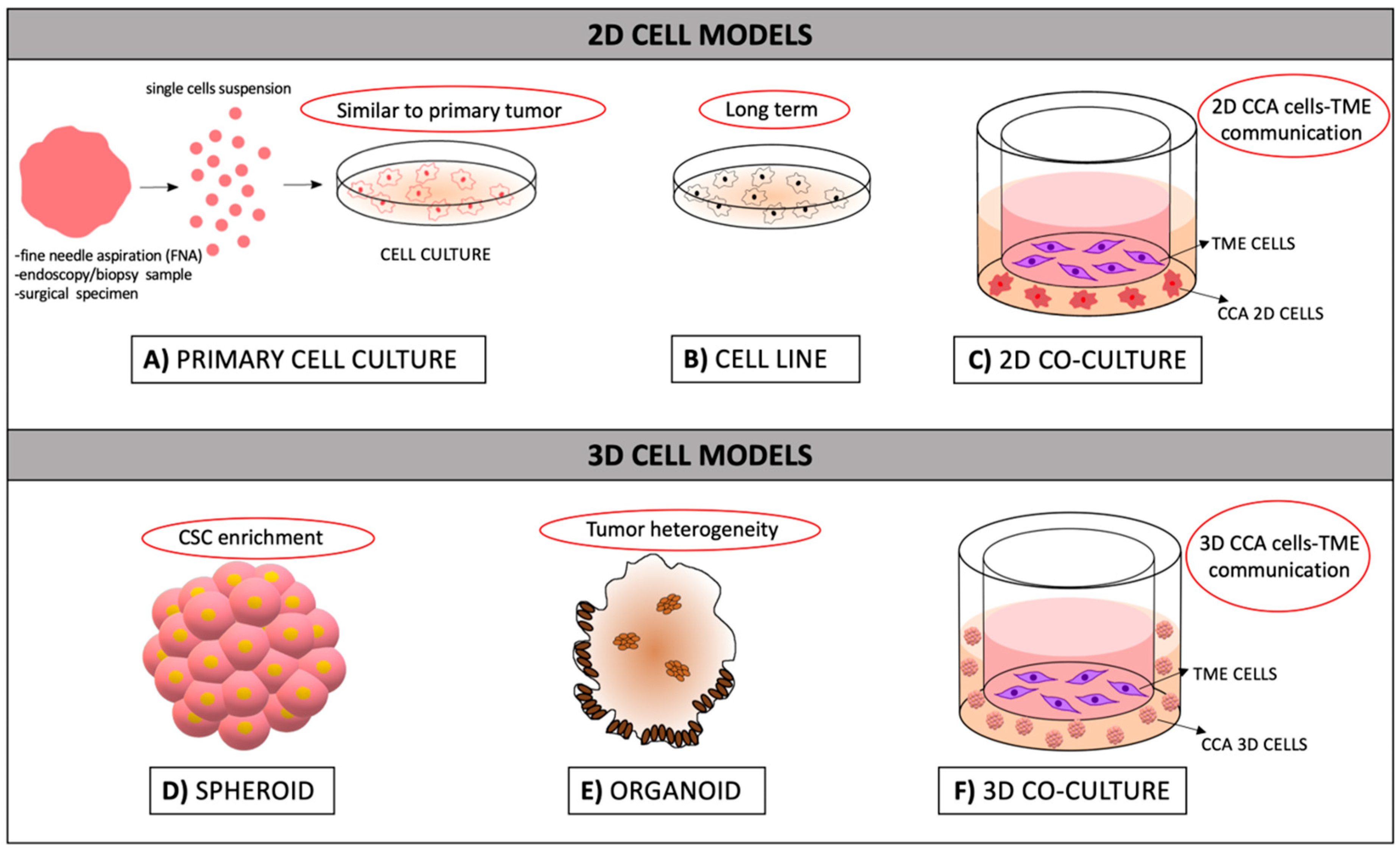
2.1. 2D Models: Cell Lines and Primary Cell Cultures
2.1.1. Strengths and Weaknesses of 2D Models
2.2. 3D CCA Modeling Approaches
2.2.1. Spheroids
2.2.2. Organoids
2.2.3. Strengths and Weaknesses of 3D Models
3. Experimental Mouse Models in CCA
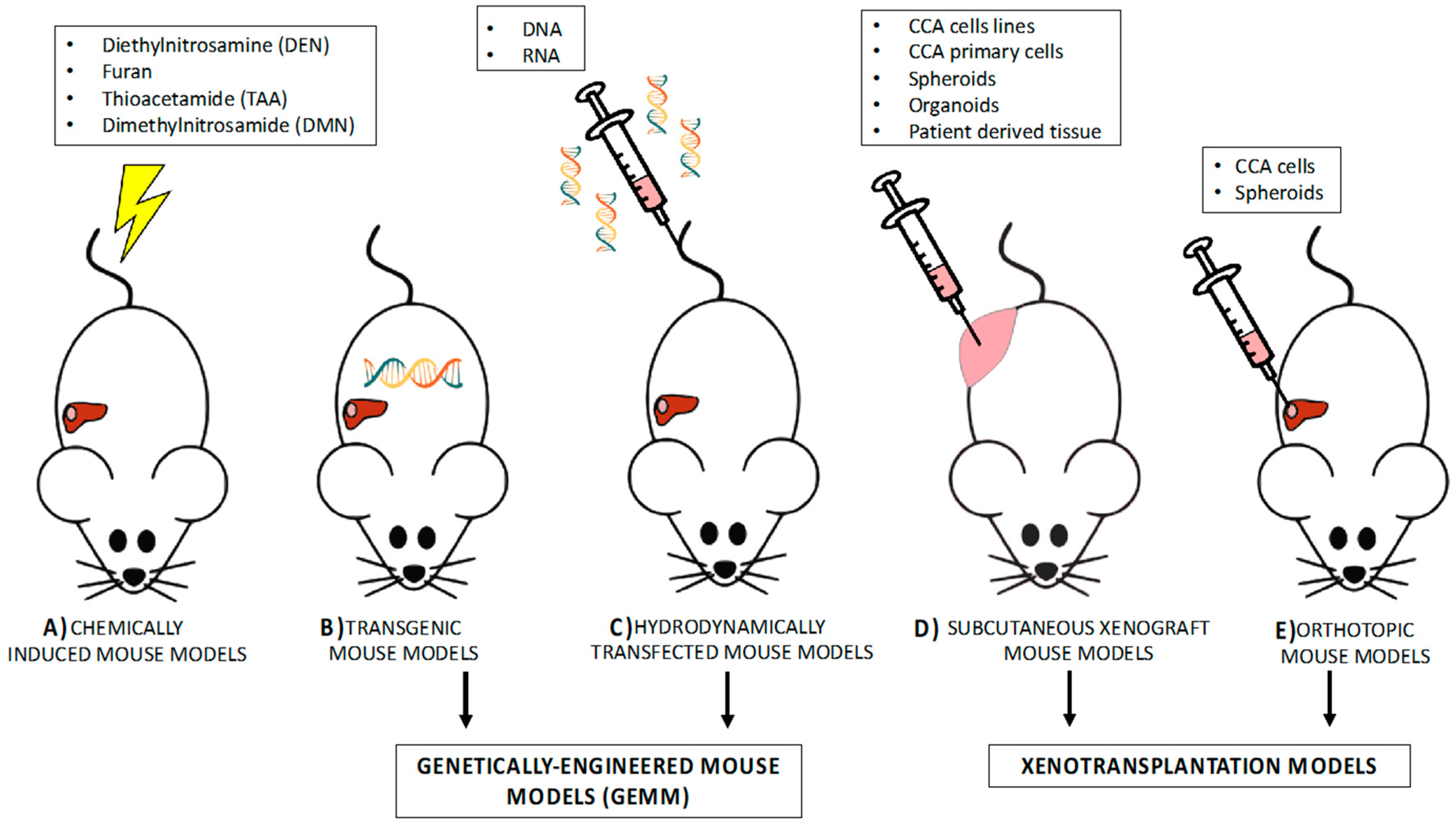
3.1. Investigating Carcinogenesis by Means of Rodent Models
3.1.1. Chemically Induced Rodent Models
3.1.2. Genetically Engineered Mouse Models (GEMM)
3.1.2.1. Transgenic Models
3.1.2.2. Hydrodynamic Transfection for Generation of Mouse Models
3.2. Experimental Mouse Models to Investigate Therapeutic Approaches in CCA
3.2.1. Ectopic Xenotransplantation Models of CCA
3.2.2. Orthotopic Mouse Models
3.2.3. Patient-Derived Xenograft (PDX)
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moazzami, B.; Majidzadeh-A, K.; Dooghaie-Moghadam, A.; Eslami, P.; Razavi-Khorasani, N.; Iravani, S.; Khoshdel, A.; Shahi, F.; Dashti, H.; Mehrvar, A.; et al. Cholangiocarcinoma: State of the Art. J. Gastrointest Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef] [Green Version]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvisé, M.; Lamarca, A. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. 1), 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39 (Suppl. 1), 19–31. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Davidson, B.R.; Goldin, R.D.; Heaton, N.; Karani, J.; Pereira, S.P.; Rosenberg, W.M.; Tait, P.; Taylor-Robinson, S.D.; Thillainayagam, A.V.; et al. Guidelines for the diagnosis and treatment of cholangiocarcinoma: An update. Gut 2012, 61, 1657–1669. [Google Scholar] [CrossRef] [Green Version]
- Alsaleh, M.; Leftley, Z.; Barbera, T.A.; Sithithaworn, P.; Khuntikeo, N.; Loilome, W.; Yongvanit, P.; Cox, I.J.; Chamodol, N.; Syms, R.R.; et al. Cholangiocarcinoma: A guide for the nonspecialist. Int. J. Gen. Med. 2019, 12, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, M.K.; Maithel, S.K. Cholangiocarcinoma: A site-specific update on the current state of surgical management and multi-modality therapy. Chin. Clin. Oncol. 2020, 9, 4. [Google Scholar] [CrossRef]
- Valle, J.W.; Furuse, J.; Jitlal, M.; Beare, S.; Mizuno, N.; Wasan, H.; Bridgewater, J.; Okusaka, T. Cisplatin and gemcitabine for advanced biliary tract cancer: A meta-analysis of two randomised trials. Ann. Oncol. 2014, 25, 391–398. [Google Scholar] [CrossRef]
- Ulahannan, S.V.; Rahma, O.E.; Duffy, A.G.; Makarova-Rusher, O.V.; Kurtoglu, M.; Liewehr, D.J.; Steinberg, S.M.; Greten, T.F. Identification of active chemotherapy regimens in advanced biliary tract carcinoma: A review of chemotherapy trials in the past two decades. Hepat. Oncol. 2015, 2, 39–50. [Google Scholar] [CrossRef]
- Yang, R.; Wang, B.; Chen, Y.J.; Li, H.B.; Hu, J.B.; Zou, S.Q. Efficacy of gemcitabine plus platinum agents for biliary tract cancers: A meta-analysis. Anticancer Drugs 2013, 24, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Weigt, J.; Malfertheiner, P. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Marin, J.J.G.; Lozano, E.; Herraez, E.; Asensio, M.; Di Giacomo, S.; Romero, M.R.; Briz, O.; Serrano, M.A.; Efferth, T.; Macias, R.I.R. Chemoresistance and chemosensitization in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1444–1453. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.; Sun, W. Targeted therapy in biliary tract cancers-current limitations and potentials in the future. J. Gastrointest Oncol. 2017, 8, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Ertel, A.; Verghese, A.; Byers, S.W.; Ochs, M.; Tozeren, A. Pathway-specific differences between tumor cell lines and normal and tumor tissue cells. Mol. Cancer 2006, 5, 55. [Google Scholar] [CrossRef] [Green Version]
- Gillet, J.P.; Calcagno, A.M.; Varma, S.; Marino, M.; Green, L.J.; Vora, M.I.; Patel, C.; Orina, J.N.; Eliseeva, T.A.; Singal, V.; et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 18708–18713. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, N.; Morioka, H.; Ohkura, H.; Hirohashi, S.; Kawai, K. Establishment and characterization of the human cholangiocarcinoma cell line HChol-Y1 in a serum-free, chemically defined medium. J. Natl. Cancer Inst. 1985, 75, 29–35. [Google Scholar]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Akita, M.; Sofue, K.; Fujikura, K.; Otani, K.; Itoh, T.; Ajiki, T.; Fukumoto, T.; Zen, Y. Histological and molecular characterization of intrahepatic bile duct cancers suggests an expanded definition of perihilar cholangiocarcinoma. HPB (Oxford) 2019, 21, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.K.; Mouradov, D.; Wasenang, W.; Luk, I.Y.; Scott, C.M.; Williams, D.S.; Yeung, Y.H.; Limpaiboon, T.; Iatropoulos, G.F.; Jenkins, L.J.; et al. Genomic Profiling of Biliary Tract Cancer Cell Lines Reveals Molecular Subtypes and Actionable Drug Targets. IScience 2019, 21, 624–637. [Google Scholar] [CrossRef] [Green Version]
- Homma, S.; Nagamori, S.; Fujise, K.; Yamazaki, K.; Hasumura, S.; Sujino, H.; Matsuura, T.; Shimizu, K.; Kameda, H.; Takaki, K. Human bile duct carcinoma cell line producing abundant mucin in vitro. Gastroenterol. Jpn. 1987, 22, 474–479. [Google Scholar] [CrossRef]
- Kusaka, Y.; Tokiwa, T.; Sato, J. Establishment and characterization of a cell line from a human cholangiocellular carcinoma. Res. Exp. Med. (Berl.) 1988, 188, 367–375. [Google Scholar] [CrossRef]
- Yeung, Y.; Lau, D.K.; Chionh, F.; Tran, H.; Tse, J.W.T.; Weickhardt, A.J.; Nikfarjam, M.; Scott, A.M.; Tebbutt, N.C.; Mariadason, J.M. K-Ras mutation and amplification status is predictive of resistance and high basal pAKT is predictive of sensitivity to everolimus in biliary tract cancer cell lines. Mol. Oncol. 2017, 11, 1130–1142. [Google Scholar] [CrossRef] [Green Version]
- Pignochino, Y.; Sarotto, I.; Peraldo-Neia, C.; Penachioni, J.Y.; Cavalloni, G.; Migliardi, G.; Casorzo, L.; Chiorino, G.; Risio, M.; Bardelli, A.; et al. Targeting EGFR/HER2 pathways enhances the antiproliferative effect of gemcitabine in biliary tract and gallbladder carcinomas. BMC Cancer 2010, 10, 631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalloni, G.; Peraldo-Neia, C.; Varamo, C.; Chiorino, G.; Sassi, F.; Aglietta, M.; Leone, F. Preclinical activity of EGFR and MEK1/2 inhibitors in the treatment of biliary tract carcinoma. Oncotarget 2016, 7, 52354–52363. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Shinbo, T.; Otagiri, H.; Saitoh, M.; Saitoh, T.; Ishizawa, S.; Shimizu, T.; Satoh, A.; Tazawa, K.; Fujimaki, M. Character of a human cholangiocarcinoma CHGS, serially transplanted to nude mice. Hum. Cell 1988, 1, 101–105. [Google Scholar] [PubMed]
- Miyagiwa, M.; Ichida, T.; Tokiwa, T.; Sato, J.; Sasaki, H. A new human cholangiocellular carcinoma cell line (HuCC-T1) producing carbohydrate antigen 19/9 in serum-free medium. In Vitro Cell. Dev. Biol. 1989, 25, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Parasramka, M.; Yan, I.K.; Wang, X.; Nguyen, P.; Matsuda, A.; Maji, S.; Foye, C.; Asmann, Y.; Patel, T. BAP1 dependent expression of long non-coding RNA NEAT-1 contributes to sensitivity to gemcitabine in cholangiocarcinoma. Mol. Cancer 2017, 16, 22. [Google Scholar] [CrossRef] [Green Version]
- Goyal, L.; Shi, L.; Liu, L.Y.; Fece de la Cruz, F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.S.; et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Vaquero, J.; Lobe, C.; Tahraoui, S.; Clapéron, A.; Mergey, M.; Merabtene, F.; Wendum, D.; Coulouarn, C.; Housset, C.; Desbois-Mouthon, C.; et al. The IGF2/IR/IGF1R Pathway in Tumor Cells and Myofibroblasts Mediates Resistance to EGFR Inhibition in Cholangiocarcinoma. Clin. Cancer Res. 2018, 24, 4282–4296. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ye, H.; Zhang, L.; Cheng, Y.; Xu, S.; Zhang, P.; Zhang, Z.; Bai, J.; Meng, F.; Zhong, L.; et al. Enhanced expression of ten-eleven translocation 1 reverses gemcitabine resistance in cholangiocarcinoma accompanied by a reduction in P-glycoprotein expression. Cancer Med. 2019, 8, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Storto, P.D.; Saidman, S.L.; Demetris, A.J.; Letessier, E.; Whiteside, T.L.; Gollin, S.M. Chromosomal breakpoints in cholangiocarcinoma cell lines. Genes Chromosomes Cancer 1990, 2, 300–310. [Google Scholar] [CrossRef]
- Sirisinha, S.; Tengchaisri, T.; Boonpucknavig, S.; Prempracha, N.; Ratanarapee, S.; Pausawasdi, A. Establishment and characterization of a cholangiocarcinoma cell line from a Thai patient with intrahepatic bile duct cancer. Asian Pac. J. Allergy Immunol. 1991, 9, 153–157. [Google Scholar]
- Intuyod, K.; Saavedra-García, P.; Zona, S.; Lai, C.F.; Jiramongkol, Y.; Vaeteewoottacharn, K.; Pairojkul, C.; Yao, S.; Yong, J.S.; Trakansuebkul, S.; et al. FOXM1 modulates 5-fluorouracil sensitivity in cholangiocarcinoma through thymidylate synthase (TYMS): Implications of FOXM1-TYMS axis uncoupling in 5-FU resistance. Cell Death Dis. 2018, 9, 1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iemura, A.; Maruiwa, M.; Yano, H.; Kojiro, M. A new human cholangiocellular carcinoma cell line (KMC-1). J. Hepatol. 1992, 15, 288–298. [Google Scholar] [CrossRef]
- Komuta, M. A new classification of primary liver carcinomas based on their possible cellular origin. J. Hepatol. 2013, 58, S266. [Google Scholar] [CrossRef]
- Shimizu, Y.; Demetris, A.J.; Gollin, S.M.; Storto, P.D.; Bedford, H.M.; Altarac, S.; Iwatsuki, S.; Herberman, R.B.; Whiteside, T.L. Two new human cholangiocarcinoma cell lines and their cytogenetics and responses to growth factors, hormones, cytokines or immunologic effector cells. Int. J. Cancer 1992, 52, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Asukai, K.; Kawamoto, K.; Eguchi, H.; Konno, M.; Asai, A.; Iwagami, Y.; Yamada, D.; Asaoka, T.; Noda, T.; Wada, H.; et al. Micro-RNA-130a-3p Regulates Gemcitabine Resistance via PPARG in Cholangiocarcinoma. Ann. Surg. Oncol. 2017, 24, 2344–2352. [Google Scholar] [CrossRef]
- Yano, H.; Iemura, A.; Haramaki, M.; Momosaki, S.; Ogasawara, S.; Higaki, K.; Kojiro, M. A human combined hepatocellular and cholangiocarcinoma cell line (KMCH-2) that shows the features of hepatocellular carcinoma or cholangiocarcinoma under different growth conditions. J. Hepatol. 1996, 24, 413–422. [Google Scholar] [CrossRef]
- Enjoji, M.; Sakai, H.; Nawata, H.; Kajiyama, K.; Tsuneyoshi, M. Sarcomatous and adenocarcinoma cell lines from the same nodule of cholangiocarcinoma. In Vitro Cell. Dev. Biol. Anim. 1997, 33, 681–683. [Google Scholar] [CrossRef]
- Saha, S.K.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef] [Green Version]
- Steffen, M.; Zuehlke, I.; Scherdin, U. Motility factors identified in supernatants of human cholangiocarcinoma cell lines. Int. J. Oncol. 2001, 18, 1107–1112. [Google Scholar] [CrossRef]
- Ku, J.L.; Yoon, K.A.; Kim, I.J.; Kim, W.H.; Jang, J.Y.; Suh, K.S.; Kim, S.W.; Park, Y.H.; Hwang, J.H.; Yoon, Y.B.; et al. Establishment and characterisation of six human biliary tract cancer cell lines. Br. J. Cancer 2002, 87, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Tepsiri, N.; Chaturat, L.; Sripa, B.; Namwat, W.; Wongkham, S.; Bhudhisawasdi, V.; Tassaneeyakul, W. Drug sensitivity and drug resistance profiles of human intrahepatic cholangiocarcinoma cell lines. World J. Gastroenterol. 2005, 11, 2748–2753. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Hu, L.; Huang, X.H.; Cao, L.Q.; Chan, K.W.; Wang, Q.; Guan, X.Y. Establishment and characterization of a human cholangiocarcinoma cell line. Oncol. Rep. 2007, 18, 1195–1200. [Google Scholar] [PubMed] [Green Version]
- Ojima, H.; Yoshikawa, D.; Ino, Y.; Shimizu, H.; Miyamoto, M.; Kokubu, A.; Hiraoka, N.; Morofuji, N.; Kondo, T.; Onaya, H.; et al. Establishment of six new human biliary tract carcinoma cell lines and identification of MAGEH1 as a candidate biomarker for predicting the efficacy of gemcitabine treatment. Cancer Sci. 2010, 101, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Obchoei, S.; Weakley, S.M.; Wongkham, S.; Wongkham, C.; Sawanyawisuth, K.; Yao, Q.; Chen, C. Cyclophilin A enhances cell proliferation and tumor growth of liver fluke-associated cholangiocarcinoma. Mol. Cancer 2011, 10, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Han, G.; Liu, H.; Qin, C. Suppression of cholangiocarcinoma cell growth by human umbilical cord mesenchymal stem cells: A possible role of Wnt and Akt signaling. PLoS ONE 2013, 8, e62844. [Google Scholar] [CrossRef] [Green Version]
- Cavalloni, G.; Peraldo-Neia, C.; Sassi, F.; Chiorino, G.; Sarotto, I.; Aglietta, M.; Leone, F. Establishment of a patient-derived intrahepatic cholangiocarcinoma xenograft model with KRAS mutation. BMC Cancer 2016, 16, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varamo, C.; Peraldo-Neia, C.; Ostano, P.; Basiricò, M.; Raggi, C.; Bernabei, P.; Venesio, T.; Berrino, E.; Aglietta, M.; Leone, F.; et al. Establishment and Characterization of a New Intrahepatic Cholangiocarcinoma Cell Line Resistant to Gemcitabine. Cancers (Basel) 2019, 11, 519. [Google Scholar] [CrossRef] [Green Version]
- Saensa-Ard, S.; Leuangwattanawanit, S.; Senggunprai, L.; Namwat, N.; Kongpetch, S.; Chamgramol, Y.; Loilome, W.; Khansaard, W.; Jusakul, A.; Prawan, A.; et al. Establishment of cholangiocarcinoma cell lines from patients in the endemic area of liver fluke infection in Thailand. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Luo, J.; Dong, X.; Yang, F.; Zhang, M.; Zhao, J.; Wang, Q.; Zhou, F.; Sun, J.; Yang, X. Establishment and Characterization of Two Novel Cholangiocarcinoma Cell Lines. Ann. Surg. Oncol. 2019, 26, 4134–4147. [Google Scholar] [CrossRef]
- Vaeteewoottacharn, K.; Pairojkul, C.; Kariya, R.; Muisuk, K.; Imtawil, K.; Chamgramol, Y.; Bhudhisawasdi, V.; Khuntikeo, N.; Pugkhem, A.; Saeseow, O.T.; et al. Establishment of Highly Transplantable Cholangiocarcinoma Cell Lines from a Patient-Derived Xenograft Mouse Model. Cells 2019, 8, 496. [Google Scholar] [CrossRef] [Green Version]
- Sato, J.; Kimura, T.; Saito, T.; Anazawa, T.; Kenjo, A.; Sato, Y.; Tsuchiya, T.; Gotoh, M. Gene expression analysis for predicting gemcitabine resistance in human cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 2011, 18, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Wattanawongdon, W.; Hahnvajanawong, C.; Namwat, N.; Kanchanawat, S.; Boonmars, T.; Jearanaikoon, P.; Leelayuwat, C.; Techasen, A.; Seubwai, W. Establishment and characterization of gemcitabine-resistant human cholangiocarcinoma cell lines with multidrug resistance and enhanced invasiveness. Int. J. Oncol. 2015, 47, 398–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, W.; Yakushiji, H.; Kitajima, Y.; Ogawa, A.; Miyazaki, K. Establishment and characterization of human hilar bile duct carcinoma cell line and cell strain. J. Hepatobiliary Pancreat. Surg. 2000, 7, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Sripa, B.; Leungwattanawanit, S.; Nitta, T.; Wongkham, C.; Bhudhisawasdi, V.; Puapairoj, A.; Sripa, C.; Miwa, M. Establishment and characterization of an opisthorchiasis-associated cholangiocarcinoma cell line (KKU-100). World J. Gastroenterol. 2005, 11, 3392–3397. [Google Scholar] [CrossRef]
- Ghosh, M.; Koike, N.; Tsunoda, S.; Hirano, T.; Kaul, S.; Kashiwagi, H.; Kawamoto, T.; Ohkohchi, N.; Saijo, K.; Ohno, T.; et al. Characterization and genetic analysis in the newly established human bile duct cancer cell lines. Int. J. Oncol. 2005, 26, 449–456. [Google Scholar] [CrossRef]
- Zach, S.; Birgin, E. Primary Cholangiocellular Carcinoma Cell Lines. J. Stem Cell Res. Transplant. 2015, 2, 1013. [Google Scholar]
- Knuth, A.; Gabbert, H.; Dippold, W.; Klein, O.; Sachsse, W.; Bitter-Suermann, D.; Prellwitz, W.; Meyer zum Büschenfelde, K.H. Biliary adenocarcinoma. Characterisation of three new human tumor cell lines. J. Hepatol. 1985, 1, 579–596. [Google Scholar] [CrossRef]
- Yoshida, K.; Tomizawa, H.; Ota, T.; Nagashima, T.; Kikuchi, H.; Watanabe, H.; Hashizaki, K.; Yonaha, A. Establishment and characterization of human cholaginocarcinoma, MEC, producing carbohydrate antigen 19-9. Hum. Cell 1990, 3, 346–351. [Google Scholar]
- Yano, H.; Maruiwa, M.; Iemura, A.; Mizoguchi, A.; Kojiro, M. Establishment and characterization of a new human extrahepatic bile duct carcinoma cell line (KMBC). Cancer 1992, 69, 1664–1673. [Google Scholar] [CrossRef]
- Saijyo, S.; Kudo, T.; Suzuki, M.; Katayose, Y.; Shinoda, M.; Muto, T.; Fukuhara, K.; Suzuki, T.; Matsuno, S. Establishment of a new extrahepatic bile duct carcinoma cell line, TFK-1. Tohoku J. Exp. Med. 1995, 177, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.Q.; Osada, M.; Ishioka, C.; Gamo, M.; Ikawa, S.; Suzuki, T.; Shimodaira, H.; Niitani, T.; Kudo, T.; Akiyama, M.; et al. Screening the p53 status of human cell lines using a yeast functional assay. Mol. Carcinog. 1997, 19, 243–253. [Google Scholar] [CrossRef]
- Kawamoto, M.; Umebayashi, M.; Tanaka, H.; Koya, N.; Nakagawa, S.; Kawabe, K.; Onishi, H.; Nakamura, M.; Morisaki, T. Combined Gemcitabine and Metronidazole Is a Promising Therapeutic Strategy for Cancer Stem-like Cholangiocarcinoma. Anticancer Res. 2018, 38, 2739–2748. [Google Scholar] [CrossRef] [Green Version]
- Yamada, N.; Chung, Y.S.; Arimoto, Y.; Sawada, T.; Seki, S.; Sowa, M. Establishment of a new human extrahepatic bile duct carcinoma cell line (OCUCh-LM1) and experimental liver metastatic model. Br. J. Cancer 1995, 71, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Han, B.; Chen, Y. Role of type IV collagenase in tumor cell invasion and effect of laminin on invasive potential in bile duct carcinoma. Zhonghua Yi Xue Za Zhi 1995, 75, 660–662, 708. [Google Scholar] [PubMed]
- Takiyama, I.; Terashima, M.; Ikeda, K.; Kawamura, H.; Kashiwaba, M.; Tamura, G.; Suto, T.; Nakashima, F.; Sasaki, R.; Saito, K. Establishment and characterization of a new human extrahepatic bile duct carcinoma cell line (ICBD-1). Oncol. Rep. 1998, 5, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Chigusa, M.; Takahashi, H.; Nakamura, J.; Tanaka, H.; Ohno, T. High level of CA19-9, CA50, and CEA-producible human cholangiocarcinoma cell line changes in the secretion ratios in vitro or in vivo. In Vitro Cell. Dev. Biol. Anim. 2000, 36, 104–109. [Google Scholar] [CrossRef]
- Kim, D.G.; Park, S.Y.; You, K.R.; Lee, G.B.; Kim, H.; Moon, W.S.; Chun, Y.H.; Park, S.H. Establishment and characterization of chromosomal aberrations in human cholangiocarcinoma cell lines by cross-species color banding. Genes Chromosomes Cancer 2001, 30, 48–56. [Google Scholar] [CrossRef]
- Yoon, H.; Min, J.K.; Lee, D.G.; Kim, D.G.; Koh, S.S.; Hong, H.J. L1 cell adhesion molecule and epidermal growth factor receptor activation confer cisplatin resistance in intrahepatic cholangiocarcinoma cells. Cancer Lett. 2012, 316, 70–76. [Google Scholar] [CrossRef]
- Rattanasinganchan, P.; Leelawat, K.; Treepongkaruna, S.A.; Tocharoentanaphol, C.; Subwongcharoen, S.; Suthiphongchai, T.; Tohtong, R. Establishment and characterization of a cholangiocarcinoma cell line (RMCCA-1) from a Thai patient. World J. Gastroenterol. 2006, 12, 6500–6506. [Google Scholar] [CrossRef]
- Murakami, T.; Yano, H.; Maruiwa, M.; Sugihara, S.; Kojiro, M. Establishment and characterization of a human combined hepatocholangiocarcinoma cell line and its heterologous transplantation in nude mice. Hepatology 1987, 7, 551–556. [Google Scholar] [CrossRef]
- Hasan, S.; Taha, R.; Omri, H.E. Current Opinions on Chemoresistance: An Overview. Bioinformation 2018, 14, 80–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.B.; Hou, Y.H.; Zhang, G.J. Correlation between EGFR mutations and serum tumor markers in lung adenocarcinoma patients. Asian Pac. J. Cancer Prev. 2013, 14, 695–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirone, G.; Shukla, A.; Marfe, G. Signaling mechanisms of resistance to EGFR- and Anti-Angiogenic Inhibitors cancer. Crit. Rev. Oncol. Hematol. 2016, 97, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Chen, Y.; Wei, L.; Li, G.; Feng, D.; Liu, S.; Jiang, R.; Zheng, S. Resistance to FGFR1-targeted therapy leads to autophagy via TAK1/AMPK activation in gastric cancer. Gastric. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Saber, A.; Haisma, H.J. CRISPR/Cas9: A powerful tool for identification of new targets for cancer treatment. Drug Discov. Today 2019, 24, 955–970. [Google Scholar] [CrossRef]
- Yoshino, J.; Akiyama, Y.; Shimada, S.; Ogura, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D.; Kudo, A.; Yamaoka, S.; et al. Loss of ARID1A induces a stemness gene ALDH1A1 expression with histone acetylation in the malignant subtype of cholangiocarcinoma. Carcinogenesis 2020, 41, 734–742. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Betge, J.; Ebert, M.P.; Boutros, M. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019, 55, 106–119. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105. [Google Scholar] [CrossRef] [Green Version]
- Kaur, G.; Dufour, J.M. Cell lines: Valuable tools or useless artifacts. Spermatogenesis 2012, 2, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Pastor, D.M.; Poritz, L.S.; Olson, T.L.; Kline, C.L.; Harris, L.R.; Koltun, W.A.; Chinchilli, V.M.; Irby, R.B. Primary cell lines: False representation or model system? a comparison of four human colorectal tumors and their coordinately established cell lines. Int. J. Clin. Exp. Med. 2010, 3, 69–83. [Google Scholar]
- Chen, Z.; Guo, P.; Xie, X.; Yu, H.; Wang, Y.; Chen, G. The role of tumour microenvironment: A new vision for cholangiocarcinoma. J. Cell. Mol. Med. 2019, 23, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Cadamuro, M.; Morton, S.D.; Strazzabosco, M.; Fabris, L. Unveiling the role of tumor reactive stroma in cholangiocarcinoma: An opportunity for new therapeutic strategies. Transl. Gastrointest Cancer 2013, 2, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Strazzabosco, M.; Crosby, H.A.; Ballardini, G.; Hubscher, S.G.; Kelly, D.A.; Neuberger, J.M.; Strain, A.J.; Joplin, R. Characterization and isolation of ductular cells coexpressing neural cell adhesion molecule and Bcl-2 from primary cholangiopathies and ductal plate malformations. Am. J. Pathol. 2000, 156, 1599–1612. [Google Scholar] [CrossRef] [Green Version]
- Holt, A.P.; Haughton, E.L.; Lalor, P.F.; Filer, A.; Buckley, C.D.; Adams, D.H. Liver myofibroblasts regulate infiltration and positioning of lymphocytes in human liver. Gastroenterology 2009, 136, 705–714. [Google Scholar] [CrossRef]
- Massani, M.; Stecca, T.; Fabris, L.; Caratozzolo, E.; Ruffolo, C.; Furlanetto, A.; Morton, S.; Cadamuro, M.; Strazzabosco, M.; Bassi, N. Isolation and characterization of biliary epithelial and stromal cells from resected human cholangiocarcinoma: A novel in vitro model to study tumor-stroma interactions. Oncol. Rep. 2013, 30, 1143–1148. [Google Scholar] [CrossRef]
- Ohira, S.; Itatsu, K.; Sasaki, M.; Harada, K.; Sato, Y.; Zen, Y.; Ishikawa, A.; Oda, K.; Nagasaka, T.; Nimura, Y.; et al. Local balance of transforming growth factor-beta1 secreted from cholangiocarcinoma cells and stromal-derived factor-1 secreted from stromal fibroblasts is a factor involved in invasion of cholangiocarcinoma. Pathol. Int. 2006, 56, 381–389. [Google Scholar] [CrossRef]
- Gentilini, A.; Rombouts, K.; Galastri, S.; Caligiuri, A.; Mingarelli, E.; Mello, T.; Marra, F.; Mantero, S.; Roncalli, M.; Invernizzi, P.; et al. Role of the stromal-derived factor-1 (SDF-1)-CXCR4 axis in the interaction between hepatic stellate cells and cholangiocarcinoma. J. Hepatol. 2012, 57, 813–820. [Google Scholar] [CrossRef]
- Chuaysri, C.; Thuwajit, P.; Paupairoj, A.; Chau-In, S.; Suthiphongchai, T.; Thuwajit, C. Alpha-smooth muscle actin-positive fibroblasts promote biliary cell proliferation and correlate with poor survival in cholangiocarcinoma. Oncol. Rep. 2009, 21, 957–969. [Google Scholar] [CrossRef] [Green Version]
- Dutta, S.; Reamtong, O.; Panvongsa, W.; Kitdumrongthum, S.; Janpipatkul, K.; Sangvanich, P.; Piyachaturawat, P.; Chairoungdua, A. Proteomics profiling of cholangiocarcinoma exosomes: A potential role of oncogenic protein transferring in cancer progression. Biochim. Biophys. Acta 2015, 1852, 1989–1999. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, Y.; Zou, L.; Zhu, Z. Role of Exosomes in Crosstalk Between Cancer-Associated Fibroblasts and Cancer Cells. Front. Oncol. 2019, 9, 356. [Google Scholar] [CrossRef]
- Li, L.; Piontek, K.; Ishida, M.; Fausther, M.; Dranoff, J.A.; Fu, R.; Mezey, E.; Gould, S.J.; Fordjour, F.K.; Meltzer, S.J.; et al. Extracellular vesicles carry microRNA-195 to intrahepatic cholangiocarcinoma and improve survival in a rat model. Hepatology 2017, 65, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Xiang, J.Y.; Ding, G.P.; Cao, L.P. Cholangiocarcinoma-derived exosomes inhibit the antitumor activity of cytokine-induced killer cells by down-regulating the secretion of tumor necrosis factor-α and perforin. J Zhejiang Univ. Sci. B 2016, 17, 537–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stock, K.; Estrada, M.F.; Vidic, S.; Gjerde, K.; Rudisch, A.; Santo, V.E.; Barbier, M.; Blom, S.; Arundkar, S.C.; Selvam, I.; et al. Capturing tumor complexity in vitro: Comparative analysis of 2D and 3D tumor models for drug discovery. Sci. Rep. 2016, 6, 28951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension: How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, K.A.; Malhotra, M.; Curtin, C.M.; O’ Brien, F.J.; O’ Driscoll, C.M. Life in 3D is never flat: 3D models to optimise drug delivery. J. Control. Release 2015, 215, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Kinney, M.A.; Hookway, T.A.; Wang, Y.; McDevitt, T.C. Engineering three-dimensional stem cell morphogenesis for the development of tissue models and scalable regenerative therapeutics. Ann. Biomed. Eng. 2014, 42, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Wang, Z.; Liu, R.; Chen, G.; Liu, L. Microfabrication-Based Three-Dimensional (3-D) Extracellular Matrix Microenvironments for Cancer and Other Diseases. Int. J. Mol. Sci. 2018, 19, 935. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Liu, R.; Jiao, Y.; Tian, C.; Farrell, J.D.; Diao, W.; Wang, X.; Zhang, F.; Yuan, W.; Han, H.; et al. A novel 3-D bio-microfluidic system mimicking in vivo heterogeneous tumour microstructures reveals complex tumour-stroma interactions. Lab Chip 2017, 17, 2852–2860. [Google Scholar] [CrossRef]
- Mao, S.; He, J.; Zhao, Y.; Liu, T.; Xie, F.; Yang, H.; Mao, Y.; Pang, Y.; Sun, W. Bioprinting of patient-derived. Biofabrication 2020. [Google Scholar] [CrossRef]
- Discher, D.E.; Janmey, P.; Wang, Y.L. Tissue cells feel and respond to the stiffness of their substrate. Science 2005, 310, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology (Bethesda) 2017, 32, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Cai, L.H.; Lan, B.; Fredberg, J.J. Hidden in the mist no more: Physical force in cell biology. Nat. Methods 2016, 13, 124–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of multicell spheroids in tissue culture as a model of nodular carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar] [PubMed]
- Zanoni, M.; Cortesi, M.; Zamagni, A.; Arienti, C.; Pignatta, S.; Tesei, A. Modeling neoplastic disease with spheroids and organoids. J. Hematol. Oncol. 2020, 13, 97. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Keysar, S.B.; Jimeno, A. More than markers: Biological significance of cancer stem cell-defining molecules. Mol. Cancer Ther. 2010, 9, 2450–2457. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, V.; Renzi, A.; Carpino, G.; Torrice, A.; Bragazzi, M.C.; Giuliante, F.; DeRose, A.M.; Fraveto, A.; Onori, P.; Napoletano, C.; et al. Profiles of cancer stem cell subpopulations in cholangiocarcinomas. Am. J. Pathol. 2015, 185, 1724–1739. [Google Scholar] [CrossRef]
- Mischiati, C.; Ura, B.; Roncoroni, L.; Elli, L.; Cervellati, C.; Squerzanti, M.; Conte, D.; Doneda, L.; Polverino de Laureto, P.; de Franceschi, G.; et al. Changes in protein expression in two cholangiocarcinoma cell lines undergoing formation of multicellular tumor spheroids in vitro. PLoS ONE 2015, 10, e0118906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G.; et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.J.; Chu, P.Y. Role of Cancer Stem Cells in Cholangiocarcinoma and Therapeutic Implications. Int. J. Mol. Sci. 2019, 20, 4154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, J.J.G.; Herraez, E.; Lozano, E.; Macias, R.I.R.; Briz, O. Models for Understanding Resistance to Chemotherapy in Liver Cancer. Cancers (Basel) 2019, 11, 1677. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, V.; Carpino, G.; Reid, L.; Gaudio, E.; Alvaro, D. Multiple cells of origin in cholangiocarcinoma underlie biological, epidemiological and clinical heterogeneity. World J. Gastrointest Oncol. 2012, 4, 94–102. [Google Scholar] [CrossRef]
- Campbell, D.J.; Dumur, C.I.; Lamour, N.F.; Dewitt, J.L.; Sirica, A.E. Novel organotypic culture model of cholangiocarcinoma progression. Hepatol. Res. 2012, 42, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, G.; Rezakhani, S.; Yildiz, E.; Nuciforo, S.; Heim, M.H.; Lutolf, M.P.; Schoonjans, K. Mechano-modulatory synthetic niches for liver organoid derivation. Nat. Commun. 2020, 11, 3416. [Google Scholar] [CrossRef]
- Grebenyuk, S.; Ranga, A. Engineering Organoid Vascularization. Front. Bioeng. Biotechnol. 2019, 7, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.J.; Chun, S.M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Saito, Y.; Nakaoka, T.; Muramatsu, T.; Ojima, H.; Sukeda, A.; Sugiyama, Y.; Uchida, R.; Furukawa, R.; Kitahara, A.; Sato, T.; et al. Induction of differentiation of intrahepatic cholangiocarcinoma cells to functional hepatocytes using an organoid culture system. Sci. Rep. 2018, 8, 2821. [Google Scholar] [CrossRef] [Green Version]
- Lampis, A.; Carotenuto, P.; Vlachogiannis, G.; Cascione, L.; Hedayat, S.; Burke, R.; Clarke, P.; Bosma, E.; Simbolo, M.; Scarpa, A.; et al. MIR21 Drives Resistance to Heat Shock Protein 90 Inhibition in Cholangiocarcinoma. Gastroenterology 2018, 154, 1066–1079.e1065. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y. Establishment of an organoid bank of biliary tract and pancreatic cancers and its application for personalized therapy and future treatment. J. Gastroenterol. Hepatol. 2019, 34, 1906–1910. [Google Scholar] [CrossRef]
- Li, M.; Wang, L.; Wang, Y.; Zhang, S.; Zhou, G.; Lieshout, R.; Ma, B.; Liu, J.; Qu, C.; Verstegen, M.M.A.; et al. Mitochondrial Fusion Via OPA1 and MFN1 Supports Liver Tumor Cell Metabolism and Growth. Cells 2020, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Koedijk, M.S.; Heijmen, B.J.M.; Groot Koerkamp, B.; Eskens, F.A.L.M.; Sprengers, D.; Poley, J.W.; van Gent, D.C.; van der Laan, L.J.W.; van der Holt, B.; Willemssen, F.E.J.A.; et al. Protocol for the STRONG trial: Stereotactic body radiation therapy following chemotherapy for unresectable perihilar cholangiocarcinoma, a phase I feasibility study. BMJ Open 2018, 8, e020731. [Google Scholar] [CrossRef] [Green Version]
- Artegiani, B.; van Voorthuijsen, L.; Lindeboom, R.G.H.; Seinstra, D.; Heo, I.; Tapia, P.; López-Iglesias, C.; Postrach, D.; Dayton, T.; Oka, R.; et al. Probing the Tumor Suppressor Function of BAP1 in CRISPR-Engineered Human Liver Organoids. Cell Stem Cell 2019, 24, 927–943.e926. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Y.; Cen, J.; Ma, X.; Cui, L.; Qiu, Z.; Zhang, Z.; Li, H.; Yang, R.Z.; Wang, C.; et al. Modelling liver cancer initiation with organoids derived from directly reprogrammed human hepatocytes. Nat. Cell Biol. 2019, 21, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Qian, M.; Chen, I.H.; Finkelstein, D.; Onar-Thomas, A.; Johnson, M.; Calabrese, C.; Bahrami, A.; López-Terrada, D.H.; Yang, J.J.; et al. Acquisition of Cholangiocarcinoma Traits during Advanced Hepatocellular Carcinoma Development in Mice. Am. J. Pathol. 2018, 188, 656–671. [Google Scholar] [CrossRef] [Green Version]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Calvo, I.; Weber, C.R.; Ray, M.; Brown, M.; Kirby, K.; Nandi, R.K.; Long, T.M.; Sparrow, S.M.; Ugolkov, A.; Qiang, W.; et al. Human Organoids Share Structural and Genetic Features with Primary Pancreatic Adenocarcinoma Tumors. Mol. Cancer Res. 2019, 17, 70–83. [Google Scholar] [CrossRef] [Green Version]
- Loeuillard, E.; Fischbach, S.R.; Gores, G.J.; Rizvi, S. Animal models of cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 982–992. [Google Scholar] [CrossRef]
- Thamavit, W.; Bhamarapravati, N.; Sahaphong, S.; Vajrasthira, S.; Angsubhakorn, S. Effects of dimethylnitrosamine on induction of cholangiocarcinoma in Opisthorchis viverrini-infected Syrian golden hamsters. Cancer Res. 1978, 38, 4634–4639. [Google Scholar]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Umemura, T.; Kai, S.; Hasegawa, R.; Kanki, K.; Kitamura, Y.; Nishikawa, A.; Hirose, M. Prevention of dual promoting effects of pentachlorophenol, an environmental pollutant, on diethylnitrosamine-induced hepato- and cholangiocarcinogenesis in mice by green tea infusion. Carcinogenesis 2003, 24, 1105–1109. [Google Scholar] [CrossRef]
- Yang, H.; Li, T.W.; Peng, J.; Tang, X.; Ko, K.S.; Xia, M.; Aller, M.A. A mouse model of cholestasis-associated cholangiocarcinoma and transcription factors involved in progression. Gastroenterology 2011, 141, 378–388.e4. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.S.; Smith, T.; Ishizaki, H.; Hong, J.Y. Enzyme mechanisms in the metabolism of nitrosamines. IARC Sci. Publ. 1991, 105, 265–274. [Google Scholar]
- Maronpot, R.R.; Giles, H.D.; Dykes, D.J.; Irwin, R.D. Furan-induced hepatic cholangiocarcinomas in Fischer 344 rats. Toxicol. Pathol. 1991, 19, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, L.W.; Sirica, A.E. Phenotypic characterization of metaplastic intestinal glands and ductular hepatocytes in cholangiofibrotic lesions rapidly induced in the caudate liver lobe of rats treated with furan. Cancer Res. 1991, 51, 5752–5759. [Google Scholar] [PubMed]
- Yeh, C.N.; Maitra, A.; Lee, K.F.; Jan, Y.Y.; Chen, M.F. Thioacetamide-induced intestinal-type cholangiocarcinoma in rat: An animal model recapitulating the multi-stage progression of human cholangiocarcinoma. Carcinogenesis 2004, 25, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Marzioni, M.; Torrice, A.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; Pierantonelli, I.; Rhönnstad, P.; Trozzi, L.; Apelqvist, T.; Gentile, R.; et al. An oestrogen receptor β-selective agonist exerts anti-neoplastic effects in experimental intrahepatic cholangiocarcinoma. Dig. Liver Dis. 2012, 44, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.B.; French, J.E.; Davis, B.J.; Haseman, J.K. The role of transgenic mouse models in carcinogen identification. Environ. Health Perspect. 2003, 111, 444–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, T.R.; Larson, M.; Wang, H.; McDermott, J.; Bronshteyn, I. Transgenic mouse technology: Principles and methods. Methods Mol. Biol. 2009, 590, 335–362. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.K.; Kim, W.H.; Jang, J.J. Expression of G1-S modulators (p53, p16, p27, cyclin D1, Rb) and Smad4/Dpc4 in intrahepatic cholangiocarcinoma. Hum. Pathol. 2002, 33, 877–883. [Google Scholar] [CrossRef]
- Downward, J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 1998, 10, 262–267. [Google Scholar] [CrossRef]
- Xu, X.; Kobayashi, S.; Qiao, W.; Li, C.; Xiao, C.; Radaeva, S.; Stiles, B.; Wang, R.H.; Ohara, N.; Yoshino, T.; et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J. Clin. Investig. 2006, 116, 1843–1852. [Google Scholar] [CrossRef]
- Ikenoue, T.; Terakado, Y.; Nakagawa, H.; Hikiba, Y.; Fujii, T.; Matsubara, D.; Noguchi, R.; Zhu, C.; Yamamoto, K.; Kudo, Y.; et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci. Rep. 2016, 6, 23899. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, M.R.; Huang, J.L.; Whitney-Miller, C.L.; Deshpande, V.; Rothberg, P.; Grose, V.; Rossi, R.M.; Zhu, A.X.; Land, H.; Bardeesy, N.; et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012, 72, 1557–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.X.; et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiguchi, K.; Carbajal, S.; Chan, K.; Beltrán, L.; Ruffino, L.; Shen, J.; Matsumoto, T.; Yoshimi, N.; DiGiovanni, J. Constitutive expression of ErbB-2 in gallbladder epithelium results in development of adenocarcinoma. Cancer Res. 2001, 61, 6971–6976. [Google Scholar]
- Yu, D.; Hung, M.C. Overexpression of ErbB2 in cancer and ErbB2-targeting strategies. Oncogene 2000, 19, 6115–6121. [Google Scholar] [CrossRef] [Green Version]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sörensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, J.J.; Zovein, A.C.; Koh, H.; Radtke, F.; Weinmaster, G.; Iruela-Arispe, M.L. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: Insights into Alagille syndrome. Development 2010, 137, 4061–4072. [Google Scholar] [CrossRef] [Green Version]
- Farazi, P.A.; Zeisberg, M.; Glickman, J.; Zhang, Y.; Kalluri, R.; DePinho, R.A. Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res. 2006, 66, 6622–6627. [Google Scholar] [CrossRef] [Green Version]
- Cadamuro, M.; Stecca, T.; Brivio, S.; Mariotti, V.; Fiorotto, R.; Spirli, C.; Strazzabosco, M.; Fabris, L. The deleterious interplay between tumor epithelia and stroma in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1435–1443. [Google Scholar] [CrossRef]
- He, L.; Tian, D.A.; Li, P.Y.; He, X.X. Mouse models of liver cancer: Progress and recommendations. Oncotarget 2015, 6, 23306–23322. [Google Scholar] [CrossRef]
- Suda, T.; Liu, D. Hydrodynamic gene delivery: Its principles and applications. Mol. Ther. 2007, 15, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Crespo, A.; Peydró, A.; Dasí, F.; Benet, M.; Calvete, J.J.; Revert, F.; Aliño, S.F. Hydrodynamic liver gene transfer mechanism involves transient sinusoidal blood stasis and massive hepatocyte endocytic vesicles. Gene Ther. 2005, 12, 927–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wang, J.; Wang, H.; Fan, L.; Fan, B.; Zeng, B.; Tao, J.; Li, X.; Che, L.; Cigliano, A.; et al. Hippo Cascade Controls Lineage Commitment of Liver Tumors in Mice and Humans. Am. J. Pathol. 2018, 188, 995–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagnaes-Hansen, F.; Holst, H.U.; Søndergaard, M.; Vorup-Jensen, T.; Flyvbjerg, A.; Jensen, U.B.; Jensen, T.G. Physiological effects of human growth hormone produced after hydrodynamic gene transfer of a plasmid vector containing the human ubiquitin promotor. J. Mol. Med. (Berl.) 2002, 80, 665–670. [Google Scholar] [CrossRef]
- Vorup-Jensen, T.; Jensen, U.B.; Liu, H.; Kawasaki, T.; Uemura, K.; Thiel, S.; Dagnaes-Hansen, F.; Jensen, T.G. Tail-vein injection of mannan-binding lectin DNA leads to high expression levels of multimeric protein in liver. Mol. Ther. 2001, 3, 867–874. [Google Scholar] [CrossRef]
- Giladi, H.; Ketzinel-Gilad, M.; Rivkin, L.; Felig, Y.; Nussbaum, O.; Galun, E. Small interfering RNA inhibits hepatitis B virus replication in mice. Mol. Ther. 2003, 8, 769–776. [Google Scholar] [CrossRef]
- Maruyama, H.; Higuchi, N.; Nishikawa, Y.; Kameda, S.; Iino, N.; Kazama, J.J.; Takahashi, N.; Sugawa, M.; Hanawa, H.; Tada, N.; et al. High-level expression of naked DNA delivered to rat liver via tail vein injection. J. Gene Med. 2002, 4, 333–341. [Google Scholar] [CrossRef]
- Zhang, G.; Gao, X.; Song, Y.K.; Vollmer, R.; Stolz, D.B.; Gasiorowski, J.Z.; Dean, D.A.; Liu, D. Hydroporation as the mechanism of hydrodynamic delivery. Gene Ther. 2004, 11, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Carlson, C.M.; Frandsen, J.L.; Kirchhof, N.; McIvor, R.S.; Largaespada, D.A. Somatic integration of an oncogene-harboring Sleeping Beauty transposon models liver tumor development in the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 17059–17064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozenne, P.; Eymin, B.; Brambilla, E.; Gazzeri, S. The ARF tumor suppressor: Structure, functions and status in cancer. Int. J. Cancer 2010, 127, 2239–2247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Song, X.; Cao, D.; Xu, Z.; Fan, B.; Che, L.; Hu, J.; Chen, B.; Dong, M.; Pilo, M.G.; et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J. Hepatol. 2017, 67, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dong, M.; Xu, Z.; Song, X.; Zhang, S.; Qiao, Y.; Che, L.; Gordan, J.; Hu, K.; Liu, Y.; et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018, 37, 3229–3242. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A.; Su, Y. Mouse xenograft models vs GEM models for human cancer therapeutics. Dis. Model Mech. 2008, 1, 78–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, C.P.; Merlino, G.; Van Dyke, T. Preclinical mouse cancer models: A maze of opportunities and challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudd, C.; Euhus, D.M.; LaRegina, M.C.; Herbold, D.R.; Palmer, D.C.; Johnson, F.E. Effect of cholecystokinin on human cholangiocarcinoma xenografted into nude mice. Cancer Res. 1985, 45, 1372–1377. [Google Scholar]
- De Minicis, S.; Kisseleva, T.; Francis, H.; Baroni, G.S.; Benedetti, A.; Brenner, D.; Alvaro, D.; Alpini, G.; Marzioni, M. Liver carcinogenesis: Rodent models of hepatocarcinoma and cholangiocarcinoma. Dig. Liver Dis. 2013, 45, 450–459. [Google Scholar] [CrossRef] [Green Version]
- Cavalloni, G.; Peraldo-Neia, C.; Sarotto, I.; Gammaitoni, L.; Migliardi, G.; Soster, M.; Marchiò, S.; Aglietta, M.; Leone, F. Antitumor activity of Src inhibitor saracatinib (AZD-0530) in preclinical models of biliary tract carcinomas. Mol. Cancer Ther. 2012, 11, 1528–1538. [Google Scholar] [CrossRef] [Green Version]
- Samukawa, E.; Fujihara, S.; Oura, K.; Iwama, H.; Yamana, Y.; Tadokoro, T.; Chiyo, T.; Kobayashi, K.; Morishita, A.; Nakahara, M.; et al. Angiotensin receptor blocker telmisartan inhibits cell proliferation and tumor growth of cholangiocarcinoma through cell cycle arrest. Int. J. Oncol. 2017, 51, 1674–1684. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Zheng, J.W.; Chen, B.; Li, H.; Li, X.; Xue, K.Y.; Ai, X.; Zou, S.Q. Effects of targeting magnetic drug nanoparticles on human cholangiocarcinoma xenografts in nude mice. Hepatobiliary Pancreat. Dis. Int. 2007, 6, 303–307. [Google Scholar]
- Zhao, X.; Zhang, C.; Zhou, H.; Xiao, B.; Cheng, Y.; Wang, J.; Yao, F.; Duan, C.; Chen, R.; Liu, Y.; et al. Synergistic antitumor activity of the combination of salubrinal and rapamycin against human cholangiocarcinoma cells. Oncotarget 2016, 7, 85492–85501. [Google Scholar] [CrossRef] [Green Version]
- Fava, G.; Marucci, L.; Glaser, S.; Francis, H.; De Morrow, S.; Benedetti, A.; Alvaro, D.; Venter, J.; Meininger, C.; Patel, T.; et al. gamma-Aminobutyric acid inhibits cholangiocarcinoma growth by cyclic AMP-dependent regulation of the protein kinase A/extracellular signal-regulated kinase 1/2 pathway. Cancer Res. 2005, 65, 11437–11446. [Google Scholar] [CrossRef] [Green Version]
- Frampton, G.A.; Lazcano, E.A.; Li, H.; Mohamad, A.; DeMorrow, S. Resveratrol enhances the sensitivity of cholangiocarcinoma to chemotherapeutic agents. Lab. Investig. 2010, 90, 1325–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, H.J.; Shim, H.E.; Han, M.E.; Kim, H.J.; Kim, K.S.; Baek, S.; Choi, K.U.; Hur, G.Y.; Oh, S.O. WTAP regulates migration and invasion of cholangiocarcinoma cells. J. Gastroenterol. 2013, 48, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Henson, R.; Braconi, C.; Patel, T. Epigallocatechin-gallate modulates chemotherapy-induced apoptosis in human cholangiocarcinoma cells. Liver Int. 2009, 29, 670–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Yamagiwa, Y.; Ueno, Y.; Patel, T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J. Hepatol. 2006, 44, 1055–1065. [Google Scholar] [CrossRef] [Green Version]
- Merino-Azpitarte, M.; Lozano, E.; Perugorria, M.J.; Esparza-Baquer, A.; Erice, O.; Santos-Laso, Á.; O’Rourke, C.J.; Andersen, J.B.; Jiménez-Agüero, R.; Lacasta, A.; et al. SOX17 regulates cholangiocyte differentiation and acts as a tumor suppressor in cholangiocarcinoma. J. Hepatol. 2017, 67, 72–83. [Google Scholar] [CrossRef]
- Yang, R.; Chen, Y.; Tang, C.; Li, H.; Wang, B.; Yan, Q.; Hu, J.; Zou, S. MicroRNA-144 suppresses cholangiocarcinoma cell proliferation and invasion through targeting platelet activating factor acetylhydrolase isoform 1b. BMC Cancer 2014, 14, 917. [Google Scholar] [CrossRef] [Green Version]
- Ursu, S.; Majid, S.; Garger, C.; de Semir, D.; Bezrookove, V.; Desprez, P.Y.; McAllister, S.; Soroceanu, L.; Nosrati, M.; Yimam, K.; et al. Novel tumor suppressor role of miRNA-876 in cholangiocarcinoma. Oncogenesis 2019, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Cadamuro, M.; Brivio, S.; Stecca, T.; Kaffe, E.; Mariotti, V.; Milani, C.; Fiorotto, R.; Spirli, C.; Strazzabosco, M.; Fabris, L. Animal models of cholangiocarcinoma: What they teach us about the human disease. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Lin, L.; Chen, C.W.; Ou, D.L. Mouse Models for Immunotherapy in Hepatocellular Carcinoma. Cancers (Basel) 2019, 11, 1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanmamed, M.F.; Chester, C.; Melero, I.; Kohrt, H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol. 2016, 27, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Fischbach, S.R.; Bronk, S.F.; Hirsova, P.; Krishnan, A.; Dhanasekaran, R.; Smadbeck, J.B.; Smoot, R.L.; Vasmatzis, G.; Gores, G.J. YAP-associated chromosomal instability and cholangiocarcinoma in mice. Oncotarget 2018, 9, 5892–5905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Boonmars, T.; Nagano, I.; Boonjaraspinyo, S.; Srinontong, P.; Ratasuwan, P.; Narong, K.; Nielsen, P.S.; Maekawa, Y. Significance of S100P as a biomarker in diagnosis, prognosis and therapy of opisthorchiasis-associated cholangiocarcinoma. Int. J. Cancer 2016, 138, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Cadamuro, M.; Spagnuolo, G.; Sambado, L.; Indraccolo, S.; Nardo, G.; Rosato, A.; Brivio, S.; Caslini, C.; Stecca, T.; Massani, M.; et al. Low-Dose Paclitaxel Reduces S100A4 Nuclear Import to Inhibit Invasion and Hematogenous Metastasis of Cholangiocarcinoma. Cancer Res. 2016, 76, 4775–4784. [Google Scholar] [CrossRef] [Green Version]
- Erice, O.; Labiano, I.; Arbelaiz, A.; Santos-Laso, A.; Munoz-Garrido, P.; Jimenez-Agüero, R.; Olaizola, P.; Caro-Maldonado, A.; Martín-Martín, N.; Carracedo, A.; et al. Differential effects of FXR or TGR5 activation in cholangiocarcinoma progression. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1335–1344. [Google Scholar] [CrossRef]
- McVeigh, L.E.; Wijetunga, I.; Ingram, N.; Marston, G.; Prasad, R.; Markham, A.F.; Coletta, P.L. Development of orthotopic tumour models using ultrasound-guided intrahepatic injection. Sci. Rep. 2019, 9, 9904. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Sveen, A.; Bruun, J.; Eide, P.W.; Eilertsen, I.A.; Ramirez, L.; Murumägi, A.; Arjama, M.; Danielsen, S.A.; Kryeziu, K.; Elez, E.; et al. Colorectal Cancer Consensus Molecular Subtypes Translated to Preclinical Models Uncover Potentially Targetable Cancer Cell Dependencies. Clin. Cancer Res. 2018, 24, 794–806. [Google Scholar] [CrossRef] [Green Version]
- Drapkin, B.J.; George, J.; Christensen, C.L.; Mino-Kenudson, M.; Dries, R.; Sundaresan, T.; Phat, S.; Myers, D.T.; Zhong, J.; Igo, P.; et al. Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts. Cancer Discov. 2018, 8, 600–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, A.A.; Xia, Y.; Trippa, L.; Le, L.P.; Igras, V.; Frederick, D.T.; Wargo, J.A.; Tanabe, K.K.; Lawrence, D.P.; Neuberg, D.S.; et al. Feasibility of Ultra-High-Throughput Functional Screening of Melanoma Biopsies for Discovery of Novel Cancer Drug Combinations. Clin. Cancer Res. 2017, 23, 4680–4692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peraldo Neia, C.; Cavalloni, G.; Chiorino, G.; Ostano, P.; Aglietta, M.; Leone, F. Gene and microRNA modulation upon trabectedin treatment in a human intrahepatic cholangiocarcinoma paired patient derived xenograft and cell line. Oncotarget 2016, 7, 86766–86780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peraldo-Neia, C.; Cavalloni, G.; Soster, M.; Gammaitoni, L.; Marchiò, S.; Sassi, F.; Trusolino, L.; Bertotti, A.; Medico, E.; Capussotti, L.; et al. Anti-cancer effect and gene modulation of ET-743 in human biliary tract carcinoma preclinical models. BMC Cancer 2014, 14, 918. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, X.; Wang, S.; Moser, C.D.; Shaleh, H.M.; Mohamed, E.A.; Chaiteerakij, R.; Allotey, L.K.; Chen, G.; Miyabe, K.; et al. Antitumor effect of FGFR inhibitors on a novel cholangiocarcinoma patient derived xenograft mouse model endogenously expressing an FGFR2-CCDC6 fusion protein. Cancer Lett. 2016, 380, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.K.; Gordan, J.D.; Kleinstiver, B.P.; Vu, P.; Najem, M.S.; Yeo, J.C.; Shi, L.; Kato, Y.; Levin, R.S.; Webber, J.T.; et al. Isocitrate Dehydrogenase Mutations Confer Dasatinib Hypersensitivity and SRC Dependence in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2016, 6, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Kabashima, A.; Hirsova, P.; Bronk, S.F.; Hernandez, M.C.; Truty, M.J.; Rizvi, S.; Kaufmann, S.H.; Gores, G.J. Fibroblast growth factor receptor inhibition induces loss of matrix MCL1 and necrosis in cholangiocarcinoma. J. Hepatol. 2018, 68, 1228–1238. [Google Scholar] [CrossRef]
- Garcia, P.L.; Miller, A.L.; Gamblin, T.L.; Council, L.N.; Christein, J.D.; Arnoletti, J.P.; Heslin, M.J.; Reddy, S.; Richardson, J.H.; Cui, X.; et al. JQ1 Induces DNA Damage and Apoptosis, and Inhibits Tumor Growth in a Patient-Derived Xenograft Model of Cholangiocarcinoma. Mol. Cancer Ther. 2018, 17, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Mancarella, S.; Serino, G.; Dituri, F.; Cigliano, A.; Ribback, S.; Wang, J.; Chen, X.; Calvisi, D.F.; Giannelli, G. Crenigacestat, a selective NOTCH1 inhibitor, reduces intrahepatic cholangiocarcinoma progression by blocking VEGFA/DLL4/MMP13 axis. Cell Death Differ. 2020, 27, 2330–2343. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.D.; Huang, C.S.; Xu, Q.C.; Li, F.; Huang, X.T.; Wang, J.Q.; Li, S.J.; Zhao, W.; Yin, X.Y. Therapeutic Targeting of CDK7 Suppresses Tumor Progression in Intrahepatic Cholangiocarcinoma. Int. J. Biol. Sci. 2020, 16, 1207–1217. [Google Scholar] [CrossRef]
- Wang, C.; Lv, H.; Yang, W.; Li, T.; Fang, T.; Lv, G.; Han, Q.; Dong, L.; Jiang, T.; Jiang, B.; et al. SVCT-2 determines the sensitivity to ascorbate-induced cell death in cholangiocarcinoma cell lines and patient derived xenografts. Cancer Lett. 2017, 398, 1–11. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cell Line/References | Anatomic Site/Classification | Source | Genetic-Molecular Alterations/References | Drug Resistance/References |
|---|---|---|---|---|
| HChol-Y1/[19] | iCCA | PT | NA | |
| Oz/[28] | iCCA | Ascites/Mts | mKRAS/[27] | |
| HuH28 a–c/[29] | iCCA | PT | mPIK3CA, mARID1A; mARID2; mMLH3; mTP53/[30]; Depmap portal | Gem a/[31] Anti-EGFR Ab c Mek inhibitors c/[32] |
| CHGS/[33] | iCCA | PT | NA | |
| HuCC-T1 c,d/[34] | iCCA | Ascites/Mts | mKRAS; iMSH6; mTP53; high level protein BAP1/ [27,35]; CCLE; Cosmic-CLP | FGFR inhibitors c/[36] EGFR inhibitors d/[37] |
| RGHuCC-T1 b | iCCA | Ascites/Mts | Gem b/[38] | |
| PCI:SG231/[39] | iCCA | PT | NA | |
| HuCCA-1 a/[40] | iCCA | PT | NA | 5-FU a/[41] |
| KMC-1/[42] | iCCA | PDX | mKRAS/[43] | |
| CC-SW-1/[44] | iCCA | PT | mBRAF/Depmap portal | |
| CC-LP-1/[44] | iCCA | PT | high level protein BAP1; mTP53; mBAP1/[35] | |
| CC-LP-1GR b | iCCA | PT | Gem b/[45] | |
| KMCH-2/[46] | iCCA-HCC | PT | NA | |
| ETK1/[47] | iCCA | Ascites, Mts | mPIK3C3; mTP53/Depmap portal | |
| RBE c/[47] | iCCA | PT | mIDH1; mBIRC6; mMSH6; mMSH3; mKRAS/ [48]; Depmap portal | FGFR inhibitors c/[36] |
| SSP-25 c/[47] | iCCA | PT | mTP53/Depmap portal | FGFR inhibitors c/[36] |
| RPMI 7451/[49] | iCCA | PT | NA | |
| SNU-1079 c/[50] | iCCA | PT | mIDH1; dARID1A; mPIK3AP1/[27]; Depmap portal | FGFR inhibitors c/[36] |
| KKU-M055/[51] | iCCA | PT | mMA2K1; high level mRNA FGFR1/[27] | |
| KKU-M156 a/[51] | iCCA | PT | NA | Cis, Carbo a/[51] |
| KKU-M214/[51] | iCCA | PT | NA | |
| KKU-M214R b | iCCA | PT | Gem, 5-FU, Doxo, PTX b/[52] | |
| KKU-OCA17/[51] | iCCA | PT | NA | |
| HKGZ-CC/[52] | iCCA | PT | mKRAS; mTP53/Depmap portal | |
| NCC-CC1/[53] | iCCA | PDX | mKRAS; mTP53/[53] | |
| NCC-CC3-1/[53] | iCCA | PDX | mKRAS/[53] | |
| NCC-CC3-2/[53] | iCCA | PDX | mKRAS/[53] | |
| NCC-CC4-1/[53] | iCCA | PDX | NA | |
| KKU-M213/[54] | iCCA | PT | mKRAS; mTP53; mSMAD4/[27]; Depmap portal | |
| HCCC-9810/[55] | iCCA | PT | NA | |
| MT-CHC01 c/[56] | iCCA | PDX | mKRAS, aErbB2, dTP53/[56] | Anti-EGFR Ab c /[32] |
| MT-CHC01R1.5 b/[57] | iCCA | PDX | Gem, 5-FU, Carbo b/[57] | |
| KKU-023/[58] | iCCA | PT | mTP53/[58] | |
| ZJU-1125/[59] | iCCA | PT | mTP53/[59] | |
| KKK-D049/[60] | iCCA | PDX | NA | |
| KKK-D068/[60] | iCCA | PDX | NA | |
| TKK/NA | iCCA | PT | high level mRNA ErbB2; aErbB2 [27] | |
| YSCCC NA | iCCA | PT | mTP53/Depmap portal | |
| YSCCCG100 b | iCCA | PT | Gem b/[61] | |
| KKU-M139/NA | iCCA | PT | NA | |
| KKU-M139R b | iCCA | PT | Gem, 5-FU, Doxo, PTX b/[62] | |
| HBDC/[63] | eCCA/pCCA | Ascites, Klatskin-Mts | NA | |
| SNU-1196/[50] | eCCA/pCCA | PT, Klatskin | aKRAS, mTP53; mSMAD6/[27,50]; Depmap portal | |
| KKU-100/[64] | eCCA/pCCA | PT, Klatskin | mKRAS; mTP53; mFGFR3/[58]; Depmap portal | |
| KKU-452/[58] | eCCA/pCCA | PT | mTP53/[58] | |
| ZJU-0826/[59] | eCCA/pCCA | PT | NA | |
| SNU-478/[50] | eCCA/dCCA | PT/Ampulla of Vater | mMLH1; mTP35/[50] | |
| SNU-869/[50] | eCCA/dCCA | PT/Ampulla of Vater | mKRAS; mPI3K, mTP53/[27,30,50] | |
| TGBC-51/[65] | eCCA/dCCA | PT/Ampulla of Vater | NA | |
| TGBC18TKB/NA | eCCA/dCCA | PT/Ampulla of Vater | mErbB2; mBRAF; high level mRNA ErbB2/[27] | |
| EGI-1 d/c/[66] | eCCA/dCCA | PT | mKRAS; mTP53/[27]; CCLE; Cosmic-CLP | EGFR inhibitors d/[37] Anti-EGFR Ab c/[32] |
| Sk-ChA-1 d/[67] | eCCA | Ascites/Mts | mBRAF; tmRASA1/[27] | EGFR inhibitors d/[37] |
| MEC/[68] | eCCA | Pleural Effusion/Mts | NA | |
| KMBC/[69] | eCCA | PT | NA | |
| TFK-1 c/[70] | eCCA/dCCA | PT | mTP53; mMSH6; mFRFR3/[71]; Depmap portal | Anti-EGFR Ab c; Mek inhibitors c/[32] |
| TFK-1GR b | eCCA/dCCA | PT | Gem b/[72] | |
| OCUCh-LM1/[73] | eCCA | Liver Mts | NA | |
| QBC939/[74] | eCCA | PT | NA | |
| RGQBC39 b | eCCA | PT | Gem b/[38] | |
| ICBD-1/[75] | eCCA | PT | NA | |
| TK/[76] | eCCA | Ascites/Mts | NA | |
| SCK/[77] | eCCA | PT | NA | |
| SCK-R b | eCCA | PT | Gem; 5-FU, Cis b/[78] | |
| JCK/[77] | eCCA | PT | NA | |
| Cho-CK/[77] | eCCA | PT | NA | |
| Choi-CK/[77] | eCCA | PT | NA | |
| SNU-245/[50] | eCCA/dCCA | PT | mBIRC6; mTP53/Depmap portal | |
| TGBC-47/[65] | eCCA | PT | NA | |
| TBCN-6/[65] | eCCA | PT | NA | |
| RMCCA-1/[79] | eCCA/pCCA | PT | NA | |
| NCC-BD1/[53] | eCCA | PDX | mKRAS; mTP53/[53] | |
| NCC-BD2/[53] | eCCA | PDX | mTP53/[53] | |
| KMCH-1 c/[80] | CCA/HCC | PT | mPTEN; dARID1A; mMSH4; mBRAF/OMICS; depmap portal | Anti-EGFR Ab; Mek inhibitors c /[32] |
| Organoid Source | Study Function | References |
|---|---|---|
| Human CCA patient | Drug screening Drug resistance Cancer differentiation Cell plasticity CCA metabolism Personalized Radiosensitivity | [135,136,137,138,139,140] |
| Human cholangiocyte GE | Cancer gene function | [141] |
| Human Hepatocytes GE | Cancer initiation Identification preventive therapies | [142] |
| Human hepatocarcinoma patient | Cancer differentiation Cell plasticity | [143,144] |
| Name | Generation | Effects | Advantages | Disadvantages | References |
|---|---|---|---|---|---|
| Smad4-Pten model | Smadco and Ptenco with Alb-cre mice. | Bile duct hyperplasia at 2 months; Tumor development at 4-7 months | Similar to human iCCA | Mixed HCC-CCA phenotype; No inflammation; No chronic liver injury; No metastases | [170] |
| KRas-IDH model | Mutant IDH2 (LSL-IDH2R172K), mice with activating KRAS mutation (LSL-KrasG12) with Alb-Cre mice. | Palpable liver tumors at 33-58 weeks | Peritoneal metastases; Similar to human iCCA | Long latency time | [48] |
| KRas-Pten model | Mice carrying a specific mutation of KRAS (LSL-KrasG12D) and/or a Ptenflox with Alb-Cre+ mice. | Multiple tumor nodules | Similar to human iCCA; Short tumor latency | No chronic liver injury; No metastases. No inflammation | [161] |
| KRas-P53 model | Alb-Cre mutants with KrasG12D mice with or without deletion of tumor protein 53. | Tumors develop at 9 weeks of age | Adjacent organ invasion; Distant metastases | No chronic liver injury; No stromal No inflammation; | [162] |
| ErbB-2A model | Bovine Keratin 5 (BK5) promoter in mice with constitutive expression of ErbB2. | Gallbladder carcinoma at 2-3 weeks;Bile duct carcinoma and iCCA at 4 months | Similar to human iCCA | Gallbladder carcinoma model; Long latency time | [164] |
| Notch1 model | Mice with overexpression of intracellular domain of Notch 1 (NICD) (Rosa26Notch1C) with Alb-Cre mice. | Changes in nuclear morphology at 8 months. | iCCA after xenotransplantation of malignant cells. | Probably mixed HCC-CCA phenotype due to the high plasticity of the transformed cells. | [166] |
| P53-/- CCL4 model | CCL4 in p53 deleted mice | Fibrosis with cholangiocyte proliferation; iCCA development in 54% of mice. | Chronic liver injury with fibrosis and inflammation. | Development of HCC; Long treatment with CCL4 | [170] |
| Tumor Type | Mice | Molecular Characterization | References |
|---|---|---|---|
| iCCA | NOD/SCID | K-Ras mutation | [56] |
| iCCA | NSG | FGFR2-CCDC6 gene fusion | [215] |
| iCCA | NGS | DH1 R132C mutation | [216] |
| iCCA | NOD/SCID | Constitutive expression of FGFR 1–4 mRNA and FRS2 | [217] |
| iCCA | CD1 immunodeficient nude | NOTCH1 overexpression | [219] |
| iCCA | B-NDG miceBALB/c (nu/nu) nude | CDK7 overexpression | [220] |
| iCCA | NOD/SCID | YAP overexpression | [204] |
| iCCA | Balb/c RJ mice | Oct-3/4 or Sox2 expression | [60] |
| iCCA | NOD/SCID | Sodium-dependent vitamin C transporter 2 (SVCT-2) expression | [221] |
| eCCA | Balb/c RJ mice | Oct-3/4 or Sox2 expression | [60] |
| Experimental Model | Advantages | Disadvantages | ||
|---|---|---|---|---|
| In vitromodels | 2 D models: | Cell lines | Long-term expansion capacity; High reproducibility of experiments; Short replication doubling time; Low maintenance costs. | Low success rate of establishing cell lines; No preservation of the cancer stem compartment; No representative of tumor genetic heterogeneity; Lack of the TME components. |
| Primary cells | Reflection of tumor heterogeneity; Preservation of the cancer stem compartment. | Short period of time to reach senescence; Low reproducibility of experiments; Lack of the TME components; Laborious to obtain. | ||
| 3 D models: | Spheroids | Recapitulation of cell interactions; Maintenance of cell physiological form and function; Reliable model for drug response assays; Enrichment of cancer stem cells population; Low production costs. | Difficult long-term maintenance; Lack TME components; Simplified architecture; Uncontrollable size and composition. | |
| Organoids | Resemble the primary tumor morphologically, genotypically and histologically; Maintenance of cell physiological form and function. In vitro model with the highest predictive patient-specific therapy response. | Low stabilization success; High production costs; Lack of tumor stroma and vascular components. | ||
| Experimental Model | Advantages | Disadvantages | ||
| In vivo models | Chemically induced models | Mimic both tumor induction and tumor progression; Identification of carcinogenic compounds; Cheap and easy to obtain. | Possible toxicity on other organs; No chance to study the role of a specific genetic mutation. | |
| Genetically engineered models | Close to human clinical condition; Spontaneous development of tumors in immunocompetent mice with an active TME; Reproduction of specific genetic aberrations of human tumors; Possibility to study cancer from its early stage. | Expensive and time-consuming; Transgenic models may have different qualitative and quantitative expression levels of integration; Tumor development is slow and variable, delay in testing therapeutic strategies. | ||
| Xenograft models: | Ectopic model | Test of therapeutic drugs; Simple protocol and easily reproducible; Lack of adverse effects in the animal; Monitor the real tumor growth by measuring the tumor mass volume. | Severe discrepancies between the origin of the tumor and the host tissue; Unphysiological implantation, rarely metastatic;Lack of immune system. | |
| Orthotopic model | Mimic the tumor milieu; Spontaneous metastases; Test of therapeutic drugs. | Time-consuming; Tumor monitoring is laborious and based on imaging tools or on animal euthanasia. | ||
| Patient Derived Xenograft | Study for personalized therapy; Retention of the stroma, 3D architecture and heterogeneity of tumor bulk. | Time-consuming; Variability in the success of engraftment. | ||
| Syngeneic model | Use of immunocompetent animals; Ideal for immunotherapeutic assessment; Test of therapeutic drugs. | Time-consuming; Biology and tumor stroma of animals, far from the human condition; Not proper for human personalized medicine. | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massa, A.; Varamo, C.; Vita, F.; Tavolari, S.; Peraldo-Neia, C.; Brandi, G.; Rizzo, A.; Cavalloni, G.; Aglietta, M. Evolution of the Experimental Models of Cholangiocarcinoma. Cancers 2020, 12, 2308. https://doi.org/10.3390/cancers12082308
Massa A, Varamo C, Vita F, Tavolari S, Peraldo-Neia C, Brandi G, Rizzo A, Cavalloni G, Aglietta M. Evolution of the Experimental Models of Cholangiocarcinoma. Cancers. 2020; 12(8):2308. https://doi.org/10.3390/cancers12082308
Chicago/Turabian StyleMassa, Annamaria, Chiara Varamo, Francesca Vita, Simona Tavolari, Caterina Peraldo-Neia, Giovanni Brandi, Alessandro Rizzo, Giuliana Cavalloni, and Massimo Aglietta. 2020. "Evolution of the Experimental Models of Cholangiocarcinoma" Cancers 12, no. 8: 2308. https://doi.org/10.3390/cancers12082308