Epithelioid Sarcoma—From Genetics to Clinical Practice

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Pathology
3. Genetics and Molecular Biology
4. Surgical Treatment
4.1. Surgical Treatment of Localized Disease
4.2. Lymphadenectomy
5. Radiotherapy
5.1. Radiotherapy of the Primary Tumor
5.2. Radiotherapy of Recurrent Disease
6. Systemic Therapy
6.1. Neoadjuvant and Adjuvant Therapy
6.2. Systemic Therapy in Advanced Disease
7. ES in Children
8. Prognosis
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Enzinger, F.M. Epithelioid sarcoma. A sarcoma simulating a granuloma or a carcinoma. Cancer 1970, 26, 1029–1041. [Google Scholar] [CrossRef]
- McPhee, M.; McGrath, B.E.; Zhang, P.; Driscoll, D.; Gibbs, J.; Peimer, C. Soft tissue sarcoma of the hand. J. Hand Surg. 1999, 24, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Alderman, A.K.; Kim, H.M.; Kotsis, S.V.; Chung, K.C. Upper-extremity sarcomas in the United States: Analysis of the surveillance, epidemiology, and end results database, 1973-1998. J. Hand Surg. 2003, 28, 511–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buecker, P.J.; Villafuerte, J.E.; Hornicek, F.J.; Gebhardt, M.C.; Mankin, H.J. Improved Survival for Sarcomas of the Wrist and Hand. J. Hand Surg. 2006, 31, 452–455. [Google Scholar] [CrossRef]
- Frezza, A.M.; Botta, L.; Pasquali, S.; Stacchiotti, S.; Gronchi, A.; Casali, P.G.; Trama, A.; Rarecarenet, W.G. 1486PAn epidemiological insight into epithelioid sarcoma (ES): The open issue of distal-type (DES) versus proximal-type (PES). Ann. Oncol. 2017, 28. [Google Scholar] [CrossRef]
- Guillou, L.; Wadden, C.; Coindre, J.-M.; Krausz, T.; Fletcher, C.D.M. Proximal-type Epithelioid Sarcoma, a Distinctive Aggressive Neoplasm Showing Rhabdoid Features: Clinicopathologic, Immunohistochemical, and Ultrastructural Study of a Series. Am. J. Surg. Pathol. 1997, 21, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Raza, M.; Verma, S.; Prasad, S.; Misra, M. Recurrent Epitheloid Cell Sarcoma of Scapular region: A Case Report and Review of Literature. J. Med. Sci. Clin. Res. 2019, 7, 160–165. [Google Scholar]
- Chbani, L.; Guillou, L.; Terrier, P.; Decouvelaere, A.V.; Gregoire, F.; Terrier-Lacombe, M.J.; Ranchere, D.; Robin, Y.M.; Collin, F.; Freneaux, P.; et al. Epithelioid sarcoma: A clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am. J. Clin. Pathol. 2009, 131, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Casanova, M.; Ferrari, A.; Collini, P.; Bisogno, G.; Alaggio, R.; Cecchetto, G.; Gronchi, A.; Meazza, C.; Garaventa, A.; Di Cataldo, A.; et al. Epithelioid sarcoma in children and adolescents: A report from the Italian Soft Tissue Sarcoma Committee. Cancer 2006, 106, 708–717. [Google Scholar] [CrossRef]
- Gross, E.; Rao, B.N.; Pappo, A.; Bowman, L.; Shearer, P.; Kaste, S.; Greenwald, C.; Michalkiewicz, E.; Pratt, C. Epithelioid sarcoma in children. J. Pediatric Surg. 1996, 31, 1663–1665. [Google Scholar] [CrossRef]
- De Viries, J.; Hoekstra, H.J.; Oosterhuis, J.W.; Postma, A.; Schraffordt Koops, H. Epithelioid sarcoma in children and adolescents: A report of four cases. J. Pediatric Surg. 1989, 24, 186–188. [Google Scholar] [CrossRef]
- Ross, H.M.; Lewis, J.J.; Woodruff, J.M.; Brennan, M.F. Epithelioid sarcoma: Clinical behavior and prognostic factors of survival. Ann. Surg. Oncol. 1997, 4, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Spillane, A.J.; Thomas, J.M.; Fisher, C. Epithelioid sarcoma: The clinicopathological complexities of this rare soft tissue sarcoma. Ann. Surg. Oncol. 2000, 7, 218–225. [Google Scholar] [CrossRef]
- Armah, H.B.; Parwani, A.V. Epithelioid Sarcoma. Arch. Pathol. Lab. Med. 2009, 133, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Thway, K.; Jones, R.L.; Noujaim, J.; Fisher, C. Epithelioid sarcoma: Diagnostic features and genetics. Adv. Anat. Pathol. 2016, 23, 41–49. [Google Scholar] [CrossRef]
- Jameson, C.F.; Simpson, M.T.; Towers, J.F. Primary epithelioid sarcoma of the hard palate. A case report. Int. J. Oral. Maxillofac. Surg. 1990, 19, 240–242. [Google Scholar] [CrossRef]
- Rossi, G.; Ferrari, G.; Longo, L.; Trentini, G.P. Epithelioid sarcoma of the penis: A case report and review of the literature. Pathol. Int. 2000, 50, 579–585. [Google Scholar] [CrossRef]
- Han, C.H.; Li, X.; Khanna, N. Epithelioid sarcoma of the vulva and its clinical implication: A case report and review of the literature. Gynecol. Oncol. Rep. 2016, 15, 31–33. [Google Scholar] [CrossRef] [Green Version]
- Chase, D.R.; Enzinger, F.M. Epithelioid sarcoma. Diagnosis, prognostic indicators, and treatment. Am. J. Surg. Pathol. 1985, 9, 241–263. [Google Scholar] [CrossRef] [Green Version]
- Bos, G.D.; Pritchard, D.J.; Reiman, H.M.; Dobyns, J.H.; Ilstrup, D.M.; Landon, G.C. Epithelioid sarcoma. An analysis of fifty-one cases. J. Bone Jt. Surg. Am. Vol. 1988, 70, 862–870. [Google Scholar] [CrossRef]
- Sakamoto, A.; Jono, O.; Hirahashi, M.; Oya, M.; Iwamoto, Y.; Arai, K. Epithelioid sarcoma with muscle metastasis detected by positron emission tomography. World J. Surg. Oncol. 2008, 6, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsamna, S.T.; Amer, K.; Elkattawy, O.; Beebe, K.S. Epithelioid sarcoma: Half a century later. Acta Oncol. 2020, 59, 48–54. [Google Scholar] [CrossRef] [PubMed]
- De Visscher, S.A.; van Ginkel, R.J.; Wobbes, T.; Veth, R.P.; Ten Heuvel, S.E.; Suurmeijer, A.J.; Hoekstra, H.J. Epithelioid sarcoma: Still an only surgically curable disease. Cancer 2006, 107, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Le Pechoux, C.; Terrier, P.; Bouaita, R.; Domont, J.; Mir, O.; Coppola, S.; Honore, C.; Le Cesne, A.; Bonvalot, S. Epithelioid sarcoma: Need for a multimodal approach to maximize the chances of curative conservative treatment. Ann. Surg. Oncol. 2014, 21, 269–276. [Google Scholar] [CrossRef]
- Prat, J.; Woodruff, J.M.; Marcove, R.C. Epithelioid sarcoma: An analysis of 22 cases indicating the prognostic significance of vascular invasion and regional lymph node metastasis. Cancer 1978, 41, 1472–1487. [Google Scholar] [CrossRef]
- Fisher, C. Epithelioid sarcoma of Enzinger. Adv. Anat. Pathol. 2006, 13, 114–121. [Google Scholar] [CrossRef]
- Kim, C.; Yoo, K.H.; Kim, M.H.; Chon, H.J.; Lee, S.I.; Lee, H.J.; Koh, S.; Lee, H.Y.; Lee, H.R.; Kim, K.S.; et al. Different subtypes of epithelioid sarcoma and their clinical implication: Long-term multi-institutional experience with a rare sarcoma. APMIS 2017, 125, 223–229. [Google Scholar] [CrossRef]
- Miettinen, M.; Fanburg-Smith, J.C.; Virolainen, M.; Shmookler, B.M.; Fetsch, J.F. Epithelioid sarcoma: An immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum. Pathol. 1999, 30, 934–942. [Google Scholar] [CrossRef]
- Evans, H.L.; Baer, S.C. Epithelioid sarcoma: A clinicopathologic and prognostic study of 26 cases. Semin. Diagn. Pathol. 1993, 10, 286–291. [Google Scholar]
- Lin, L.; Hicks, D.; Xu, B.; Sigel, J.E.; Bergfeld, W.F.; Montgomery, E.; Fisher, C.; Hartke, M.; Tubbs, R.; Goldblum, J.R. Expression profile and molecular genetic regulation of cyclin D1 expression in epithelioid sarcoma. Mod. Pathol. 2005, 18, 705–709. [Google Scholar] [CrossRef] [Green Version]
- Tsakonas, G.P.; Kallistratos, M.S.; Balamoti, E.K.; Gassiamis, A.; Zizi-Sermpetzoglou, A.; Mylonakis, N.; Karabelis, A.; Kosmas, C. Rare and aggressive metastatic, axial multifocal local epithelioid sarcoma associated with paraneoplastic granulocytosis and hypoglycaemia. Lancet Oncol. 2007, 8, 82–84. [Google Scholar] [CrossRef]
- Kato, H.; Hatori, M.; Kokubun, S.; Watanabe, M.; Smith, R.A.; Hotta, T.; Ogose, A.; Morita, T.; Murakami, T.; Aiba, S. CA125 Expression in Epithelioid Sarcoma. Jpn. J. Clin. Oncol. 2004, 34, 149–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, M.; Kawashima, H.; Ogose, A.; Kudo, N.; Ariizumi, T.; Hotta, T.; Umezu, H.; Hatano, H.; Morita, T.; Nishio, J.; et al. Serum CA 125 expression as a tumor marker for diagnosis and monitoring the clinical course of epithelioid sarcoma. J. Cancer Res. Clin. Oncol. 2010, 136, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Manivel, J.C.; Wick, M.R.; Dehner, L.P.; Sibley, R.K. Epithelioid Sarcoma: An Immunohistochemical Study. Am. J. Clin. Pathol. 1987, 87, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Stockman, D.L.; Hornick, J.L.; Deavers, M.T.; Lev, D.C.; Lazar, A.J.; Wang, W.-L. ERG and FLI1 protein expression in epithelioid sarcoma. Mod. Pathol. 2014, 27, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Rakheja, D.; Wilson, K.S.; Meehan, J.; Schultz, R.A.; Gomez, A.M. Proximal-type and Classic Epithelioid Sarcomas Represent a Clinicopathologic Continuum: Case Report. Pediatric Dev. Pathol. 2005, 8, 105–114. [Google Scholar] [CrossRef]
- Hornick, J.L.; Dal Cin, P.; Fletcher, C.D.M. Loss of INI1 Expression is Characteristic of Both Conventional and Proximal-type Epithelioid Sarcoma. Am. J. Surg. Pathol. 2009, 33, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Siontis, B.L.; Chugh, R.; Schuetze, S.M. The potential of emerging therapeutics for epithelioid sarcoma. Expert Opin. Orphan Drugs 2017, 5, 983–989. [Google Scholar] [CrossRef]
- Brenca, M.; Rossi, S.; Lorenzetto, E.; Piccinin, E.; Piccinin, S.; Rossi, F.M.; Giuliano, A.; Dei Tos, A.P.; Maestro, R.; Modena, P. SMARCB1/INI1 genetic inactivation is responsible for tumorigenic properties of epithelioid sarcoma cell line VAESBJ. Mol. Cancer Ther. 2013, 12, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Kohashi, K.; Izumi, T.; Oda, Y.; Yamamoto, H.; Tamiya, S.; Taguchi, T.; Iwamoto, Y.; Hasegawa, T.; Tsuneyoshi, M. Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: A useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum. Pathol. 2009, 40, 349–355. [Google Scholar] [CrossRef]
- Sullivan, L.M.; Folpe, A.L.; Pawel, B.R.; Judkins, A.R.; Biegel, J.A. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod. Pathol. 2013, 26, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Papp, G.; Changchien, Y.-C.; Péterfia, B.; Pecsenka, L.; Krausz, T.; Stricker, T.P.; Khoor, A.; Donner, L.; Sápi, Z. SMARCB1 protein and mRNA loss is not caused by promoter and histone hypermethylation in epithelioid sarcoma. Mod. Pathol. 2013, 26, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallcup, M.R. Role of protein methylation in chromatin remodeling and transcriptional regulation. Oncogene 2001, 20, 3014–3020. [Google Scholar] [CrossRef] [Green Version]
- Clair, J.M.-S.; Soydaner-Azeloglu, R.; Lee, K.E.; Taylor, L.; Livanos, A.; Pylayeva-Gupta, Y.; Miller, G.; Margueron, R.; Reinberg, D.; Bar-Sagi, D. EZH2 couples pancreatic regeneration to neoplastic progression. Genes Dev. 2012, 26, 439–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, A. Role of the EZH2 histone methyltransferase as a therapeutic target in cancer. Pharmacol. Ther. 2016, 165, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Porter Scott, M.; Chesworth, R.; Moyer, M.P.; Copeland, R.A.; et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, G.; Song, Y.; Lam, R.; Ruder, D.; Creighton, C.J.; Bid, H.K.; Bill, K.L.; Bolshakov, S.; Zhang, X.; Lev, D.; et al. HDAC Inhibition for the Treatment of Epithelioid Sarcoma: Novel Cross Talk Between Epigenetic Components. Mol. Cancer Res. 2016, 14, 35. [Google Scholar] [CrossRef] [Green Version]
- RUIJTER, A.J.M.d.; GENNIP, A.H.v.; CARON, H.N.; KEMP, S.; KUILENBURG, A.B.P.v. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Schoffski, P.; Jones, R.; Agulnik, M.; Villalobos, V.M.; Jahan, T.M.; Chen, T.W.-W.; Italiano, A.; Demetri, G.D.; Cote, G.M.; et al. Safety and efficacy of tazemetostat, a first-in-class EZH2 inhibitor, in patients (pts) with epithelioid sarcoma (ES) (NCT02601950). J. Clin. Oncol. 2019, 37, 11003. [Google Scholar] [CrossRef]
- Xie, X.; Ghadimi, M.P.H.; Young, E.D.; Belousov, R.; Zhu, Q.-s.; Liu, J.; Lopez, G.; Colombo, C.; Peng, T.; Reynoso, D.; et al. Combining EGFR and mTOR Blockade for the Treatment of Epithelioid Sarcoma. Clin. Cancer Res. 2011, 17, 5901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darr, J.; Klochendler, A.; Isaac, S.; Eden, A. Loss of IGFBP7 expression and persistent AKT activation contribute to SMARCB1/Snf5-mediated tumorigenesis. Oncogene 2014, 33, 3024–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imura, Y.; Yasui, H.; Outani, H.; Wakamatsu, T.; Hamada, K.; Nakai, T.; Yamada, S.; Myoui, A.; Araki, N.; Ueda, T.; et al. Combined targeting of mTOR and c-MET signaling pathways for effective management of epithelioid sarcoma. Mol. Cancer 2014, 13, 185. [Google Scholar] [CrossRef] [Green Version]
- Cascio, M.J.; O’Donnell, R.J.; Horvai, A.E. Epithelioid sarcoma expresses epidermal growth factor receptor but gene amplification and kinase domain mutations are rare. Mod. Pathol. 2010, 23, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumors Are Angiogenesis Dependent? J. Natl. Cancer Inst. JNCI. 1990, 82, 4. [Google Scholar] [CrossRef] [PubMed]
- Kuhnen, C.; Lehnhardt, M.; Tolnay, E.; Muehlberger, T.; Vogt, P.M.; Müller, K.-M. Patterns of expression and secretion of vascular endothelial growth factor in malignant soft-tissue tumours. J. Cancer Res. Clin. Oncol. 2000, 126, 219–225. [Google Scholar] [CrossRef]
- Sakharpe, A.; Lahat, G.; Gulamhusein, T.; Liu, P.; Bolshakov, S.; Nguyen, T.; Zhang, P.; Belousov, R.; Young, E.; Xie, X.; et al. Epithelioid sarcoma and unclassified sarcoma with epithelioid features: Clinicopathological variables, molecular markers, and a new experimental model. Oncologist 2011, 16, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.; Zheng, G. E-cadherin/beta-catenin complex and the epithelial barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef] [Green Version]
- Ino, Y.; Gotoh, M.; Sakamoto, M.; Tsukagoshi, K.; Hirohashi, S. Dysadherin, a cancer-associated cell membrane glycoprotein, down-regulates E-cadherin and promotes metastasis. Proc. Natl. Acad. Sci. USA 2002, 99, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Izumi, T.; Oda, Y.; Hasegawa, T.; Nakanishi, Y.; Iwasaki, H.; Sonobe, H.; Goto, H.; Kusakabe, H.; Takahira, T.; Kobayashi, C.; et al. Prognostic significance of dysadherin expression in epithelioid sarcoma and its diagnostic utility in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Mod. Pathol. 2006, 19, 820–831. [Google Scholar] [CrossRef]
- Frezza, A.M.; Sigalotti, L.; Del Savio, E.; Baldazzi, D.; Sbaraglia, M.; Righi, A.; Gambarotti, M.; Barisella, M.; Brich, S.; Dei Tos, A.P.; et al. Epitheliod sarcoma: Molecular insights into proximal versus classic variant. J. Clin. Oncol. 2020, 38, e23552. [Google Scholar] [CrossRef]
- Noujaim, J.; Thway, K.; Bajwa, Z.; Bajwa, A.; Maki, R.G.; Jones, R.L.; Keller, C. Epithelioid Sarcoma: Opportunities for Biology-Driven Targeted Therapy. Front. Oncol. 2015, 5, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.W.; Jee, K.J.; Han, S.S.; Gong, G.Y.; Choi, J.H.; Moon, K.C.; Koh, J.K. Comparative genomic hybridization in epithelioid sarcoma. Br. J. Derm. 2004, 151, 1054–1059. [Google Scholar] [CrossRef]
- Emori, M.; Tsukahara, T.; Murase, M.; Kano, M.; Murata, K.; Takahashi, A.; Kubo, T.; Asanuma, H.; Yasuda, K.; Kochin, V. High expression of CD109 antigen regulates the phenotype of cancer stem-like cells/cancer-initiating cells in the novel epithelioid sarcoma cell line ESX and is related to poor prognosis of soft tissue sarcoma. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- De Vita, A.; Recine, F.; Mercatali, L.; Miserocchi, G.; Liverani, C.; Spadazzi, C.; Casadei, R.; Bongiovanni, A.; Pieri, F.; Riva, N.; et al. Myxofibrosarcoma primary cultures: Molecular and pharmacological profile. Ther. Adv. Med. Oncol. 2017, 9, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miserocchi, G.; Mercatali, L.; Liverani, C.; De Vita, A.; Spadazzi, C.; Pieri, F.; Bongiovanni, A.; Recine, F.; Amadori, D.; Ibrahim, T. Management and potentialities of primary cancer cultures in preclinical and translational studies. J. Transl. Med. 2017, 15, 229. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, A.; Grimer, R.J.; Abudu, A.; Tillman, R.M.; Carter, S.R.; Jeys, L.; Ferguson, P.C.; Griffin, A.M.; Wunder, J.S. Epithelioid sarcomas: How important is locoregional control? Eur. J. Surg. Oncol. 2017, 43, 1746–1752. [Google Scholar] [CrossRef]
- Rutkowski, P. Soft Tissue Sarcomas; Rutkowski, P., Ed.; Via Medica: Gdansk, Poland, 2016. [Google Scholar]
- Matsushita, Y.; Ahmed, A.R.; Kawaguchi, N.; Matsumoto, S.; Manabe, J. Epithelioid sarcoma of the extremities: A dismal long-term outcome. J. Orthop. Sci. 2002, 7, 462–466. [Google Scholar] [CrossRef]
- Guzzetta, A.A.; Montgomery, E.A.; Lyu, H.; Hooker, C.M.; Meyer, C.F.; Loeb, D.M.; Frassica, D.; Weber, K.L.; Ahuja, N. Epithelioid sarcoma: One institution’s experience with a rare sarcoma. J. Surg. Res. 2012, 177, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Baratti, D.; Pennacchioli, E.; Casali, P.G.; Bertulli, R.; Lozza, L.; Olmi, P.; Collini, P.; Radaelli, S.; Fiore, M.; Gronchi, A. Epithelioid sarcoma: Prognostic factors and survival in a series of patients treated at a single institution. Ann. Surg. Oncol. 2007, 14, 3542–3551. [Google Scholar] [CrossRef]
- Livi, L.; Shah, N.; Paiar, F.; Fisher, C.; Judson, I.; Moskovic, E.; Thomas, M.; Harmer, C. Treatment of epithelioid sarcoma at the Royal Marsden Hospital. Sarcoma 2003, 7, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, J.J.; Tan, C.C.; Casper, E.S.; Collin, C.F.; Friedrich, C.; Shiu, M.; Hajdu, S.I.; Brennan, M.F. Refinement of clinicopathologic staging for localized soft tissue sarcoma of the extremity: A study of 423 adults. J. Clin. Oncol. 1992, 10, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Yoshida, A.; Ogura, K.; Kobayashi, E.; Susa, M.; Morioka, H.; Iwata, S.; Ishii, T.; Hiruma, T.; Chuman, H.; et al. Prognostic Value of Relevant Clinicopathologic Variables in Epithelioid Sarcoma: A Multi-Institutional Retrospective Study of 44 Patients. Ann. Surg. Oncol. 2015, 22, 2624–2632. [Google Scholar] [CrossRef] [Green Version]
- Keung, E.Z.; Chiang, Y.J.; Voss, R.K.; Cormier, J.N.; Torres, K.E.; Hunt, K.K.; Feig, B.W.; Roland, C.L. Defining the incidence and clinical significance of lymph node metastasis in soft tissue sarcoma. Eur. J. Surg. Oncol. 2018, 44, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Daigeler, A.; Kuhnen, C.; Moritz, R.; Stricker, I.; Goertz, O.; Tilkorn, D.; Steinstraesser, L.; Steinau, H.U.; Lehnhardt, M. Lymph node metastases in soft tissue sarcomas: A single center analysis of 1597 patients. Langenbecks Arch. Surg. 2009, 394, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Andreou, D.; Boldt, H.; Werner, M.; Hamann, C.; Pink, D.; Tunn, P.U. Sentinel node biopsy in soft tissue sarcoma subtypes with a high propensity for regional lymphatic spread—results of a large prospective trial. Ann. Oncol. 2013, 24, 1400–1405. [Google Scholar] [CrossRef]
- Sobanko, J.F.; Meijer, L.; Nigra, T.P. Epithelioid sarcoma: A review and update. J. Clin. Aesthet. Derm. 2009, 2, 49–54. [Google Scholar]
- Maduekwe, U.N.; Hornicek, F.J.; Springfield, D.S.; Raskin, K.A.; Harmon, D.C.; Choy, E.; Rosenberg, A.E.; Nielsen, G.P.; DeLaney, T.F.; Chen, Y.L.; et al. Role of sentinel lymph node biopsy in the staging of synovial, epithelioid, and clear cell sarcomas. Ann. Surg. Oncol. 2009, 16, 1356–1363. [Google Scholar] [CrossRef]
- Seal, A.; Tse, R.; Wehrli, B.; Hammond, A.; Temple, C.L. Sentinel node biopsy as an adjunct to limb salvage surgery for epithelioid sarcoma of the hand. World J. Surg. Oncol. 2005, 3, 41. [Google Scholar] [CrossRef] [Green Version]
- Blazer, D.G., 3rd; Sabel, M.S.; Sondak, V.K. Is there a role for sentinel lymph node biopsy in the management of sarcoma? Surg. Oncol. 2003, 12, 201–206. [Google Scholar] [CrossRef]
- Callister, M.D.; Ballo, M.T.; Pisters, P.W.T.; Patel, S.R.; Feig, B.W.; Pollock, R.E.; Benjamin, R.S.; Zagars, G.K. Epithelioid sarcoma: Results of conservative surgery and radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 384–391. [Google Scholar] [CrossRef]
- Haas, R.L.M.; Miah, A.B.; LePechoux, C.; DeLaney, T.F.; Baldini, E.H.; Alektiar, K.; O’Sullivan, B. Preoperative radiotherapy for extremity soft tissue sarcoma; past, present and future perspectives on dose fractionation regimens and combined modality strategies. Radiother. Oncol. 2016, 119, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, P.S.; Flum, D.R.; Tanas, M.R.; Rubin, B.P.; Mann, G.N. Epithelioid sarcoma: The University of Washington experience. Am. J. Surg. 2008, 196, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Shimm, D.S.; Suit, H.D. Radiation therapy of epithelioid sarcoma. Cancer 1983, 52, 1022–1025. [Google Scholar] [CrossRef]
- Casali, P.G.; Abecassis, N.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; Brodowicz, T.; Broto, J.M.; et al. Soft tissue and visceral sarcomas: ESMO–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv51–iv67. [Google Scholar] [CrossRef]
- Naghavi, A.O.; Fernandez, D.C.; Mesko, N.; Juloori, A.; Martinez, A.; Scott, J.G.; Shah, C.; Harrison, L.B. American Brachytherapy Society consensus statement for soft tissue sarcoma brachytherapy. Brachytherapy 2017, 16, 466–489. [Google Scholar] [CrossRef]
- De Jong, M.A.A.; Oldenborg, S.; Bing Oei, S.; Griesdoorn, V.; Kolff, M.W.; Koning, C.C.E.; van Tienhoven, G. Reirradiation and hyperthermia for radiation-associated sarcoma. Cancer 2012, 118, 180–187. [Google Scholar] [CrossRef]
- Stragliotto, C.L.; Karlsson, K.; Lax, I.; Rutkowska, E.; Bergh, J.; Strander, H.; Blomgren, H.; Friesland, S. A retrospective study of SBRT of metastases in patients with primary sarcoma. Med. Oncol. (Northwood, London, England) 2012, 29, 3431–3439. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, A.D.; Haupt, E.E.; Chan, C.M.; Spiguel, A.R.; Scarborough, M.T.; Zlotecki, R.A.; Gibbs, P.C. Treatment of Sarcoma Lung Metastases with Stereotactic Body Radiotherapy. Sarcoma 2018, 2018, 9132359. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.L.; Constantinidou, A.; Olmos, D.; Thway, K.; Fisher, C.; Al-Muderis, O.; Scurr, M.; Judson, I.R. Role of palliative chemotherapy in advanced epithelioid sarcoma. Am. J. Clin. Oncol. 2012, 35, 351–357. [Google Scholar] [CrossRef]
- Rekhi, B.; Gorad, B.D.; Chinoy, R.F. Clinicopathological features with outcomes of a series of conventional and proximal-type epithelioid sarcomas, diagnosed over a period of 10 years at a tertiary cancer hospital in India. Virchows Arch. 2008, 453, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Outani, H.; Imura, Y.; Tanaka, T.; Takenaka, S.; Oshima, K.; Hamada, K.; Kakunaga, S.; Joyama, S.; Naka, N.; Kudawara, I.; et al. Clinical outcomes of patients with epithelioid sarcomas: Impact and management of nodal metastasis. Int. J. Clin. Oncol. 2018, 23, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Frezza, A.M.; Jones, R.L.; Lo Vullo, S.; Asano, N.; Lucibello, F.; Ben-Ami, E.; Ratan, R.; Teterycz, P.; Boye, K.; Brahmi, M.; et al. Anthracycline, Gemcitabine, and Pazopanib in Epithelioid Sarcoma: A Multi-institutional Case Series. JAMA Oncol. 2018, 4, e180219. [Google Scholar] [CrossRef] [PubMed]
- Pink, D.; Richter, S.; Gerdes, S.; Andreou, D.; Tunn, P.U.; Busemann, C.; Ehninger, G.; Reichardt, P.; Schuler, M.K. Gemcitabine and docetaxel for epithelioid sarcoma: Results from a retrospective, multi-institutional analysis. Oncology 2014, 87, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Touati, N.; Schoffski, P.; Litiere, S.; Judson, I.; Sleijfer, S.; van der Graaf, W.T.; Italiano, A.; Isambert, N.; Gil, T.; Blay, J.Y.; et al. European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Experience with Advanced/Metastatic Epithelioid Sarcoma Patients Treated in Prospective Trials: Clinical Profile and Response to Systemic Therapy. Clin. Oncol. (R Coll. Radiol.) 2018, 30, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Tariq, Z.; Ghose, A.; Bawany, M.Z.; Saeed, B.; Mohamed, I.; Harmon, D. A case report of complete remission of pulmonary metastases from epithelioid sarcoma to navelbine chemotherapy. Am. J. Ther. 2012, 19, e95–e97. [Google Scholar] [CrossRef]
- Anderson, S.E.; Keohan, M.L.; D’Adamo, D.R.; Maki, R.G. A retrospective analysis of vinorelbine chemotherapy for patients with previously treated soft-tissue sarcomas. Sarcoma 2006, 2006, 15947. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Matsumine, A.; Kawai, A.; Araki, N.; Goto, T.; Yonemoto, T.; Sugiura, H.; Nishida, Y.; Hiraga, H.; Honoki, K.; et al. The clinical outcome of pazopanib treatment in Japanese patients with relapsed soft tissue sarcoma: A Japanese Musculoskeletal Oncology Group (JMOG) study. Cancer 2016, 122, 1408–1416. [Google Scholar] [CrossRef]
- Irimura, S.; Nishimoto, K.; Kikuta, K.; Nakayama, R.; Susa, M.; Horiuchi, K.; Nakamura, M.; Matsumoto, M.; Morioka, H. Successful Treatment with Pazopanib for Multiple Lung Metastases of Inguinal Epithelioid Sarcoma: A Case Report. Case Rep. Oncol. 2015, 8, 378–384. [Google Scholar] [CrossRef]
- Gounder, M.M.; Stacchiotti, S.; Schoffski, P.; Cote, G.M.; Villalobos, V.M.; Jahan, T.M.; Chen, T.W.-W.; Ratan, R.; Gupta, A.A.; Dileo, P.; et al. Efficacy, safety, and immune priming effect of tazemetostat in patients with epithelioid sarcoma. J. Clin. Oncol. 2020, 38, 11564. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.-C.; Toulmonde, M.; Michot, J.-M.; Lucchesi, C.; Varga, A.; Coindre, J.-M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Sen, S.; McKean, M.A.; Sierra, L.; Ainscough, J.; Yang, J.; Hamlett, A.; Zimmerman, T.; Merchant, M.; Chawla, S.P. A phase Ib/III randomized, double-blind, placebo-controlled study of tazemetostat plus doxorubicin as frontline therapy for patients with advanced epithelioid sarcoma. J. Clin. Oncol. 2020, 38, TPS11573. [Google Scholar] [CrossRef]
- Schuetze, S.M.; Bolejack, V.; Choy, E.; Ganjoo, K.N.; Staddon, A.P.; Chow, W.A.; Tawbi, H.A.; Samuels, B.L.; Patel, S.R.; von Mehren, M.; et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer 2017, 123, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Penot, P.; Bouaziz, J.D.; Battistella, M.; Kerob, D.; Pages, C.; Vilmer, C.; Basset-Seguin, N.; Madjessli, N.; Comte, C.; Farges, C.; et al. Stabilization of multiple metastatic epithelioid sarcoma under treatment with sunitinib malate. Br. J. Derm. 2013, 168, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Kang, H.J.; Yalon-Oren, M.; Marshall, L.V.; Vezina, C.; Pappo, A.; Laetsch, T.W.; Petrilli, A.S.; Ebinger, M.; Toporski, J.; et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): Interim analysis of an open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 121–133. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.; Rosen, G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Spunt, S.L.; Million, L.; Chi, Y.-Y.; Anderson, J.; Tian, J.; Hibbitts, E.; Coffin, C.; McCarville, M.B.; Randall, R.L.; Parham, D.M.; et al. A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): A Children’s Oncology Group prospective study. Lancet Oncol. 2020, 21, 145–161. [Google Scholar] [CrossRef]
- Spunt, S.L.; Francotte, N.; De Salvo, G.L.; Chi, Y.Y.; Zanetti, I.; Hayes-Jordan, A.; Kao, S.C.; Orbach, D.; Brennan, B.; Weiss, A.R.; et al. Clinical features and outcomes of young patients with epithelioid sarcoma: An analysis from the Children’s Oncology Group and the European paediatric soft tissue Sarcoma Study Group prospective clinical trials. Eur. J. Cancer. 2019, 112, 98–106. [Google Scholar] [CrossRef]
- Sparber-Sauer, M.; Koscielniak, E.; Vokuhl, C.; Seitz, G.; Hallmen, E.; von Kalle, T.; Scheer, M.; Munter, M.; Bielack, S.S.; Ladenstein, R.; et al. Epithelioid sarcoma in children, adolescents, and young adults: Localized, primary metastatic and relapsed disease. Treatment results of five Cooperative Weichteilsarkom Studiengruppe (CWS) trials and one registry. Pediatr. Blood Cancer 2019, 66, e27879. [Google Scholar] [CrossRef]
- Gupta, H.; Davidoff, A.M.; Rao, B.N.; Jenkins, J.J.; Spunt, S.L. Neonatal epithelioid sarcoma: A distinct clinical entity? J. Pediatr. Surg. 2006, 41, e9–e11. [Google Scholar] [CrossRef]
- Ferrari, A.; Casanova, M.; Collini, P.; Meazza, C.; Luksch, R.; Massimino, M.; Cefalo, G.; Terenziani, M.; Spreafico, F.; Catania, S.; et al. Adult-type soft tissue sarcomas in pediatric-age patients: Experience at the Istituto Nazionale Tumori in Milan. J. Clin. Oncol. 2005, 23, 4021–4030. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Facchinetti, F.; Boeri, M.; Lorenzetto, E.; Livio, A.; Gronchi, A.; Ferrari, A.; Massimino, M.; Spreafico, F.; Giangaspero, F. Prognostic determinants in epithelioid sarcoma. Eur. J. Cancer 2011, 47, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Jawad, M.U.; Extein, J.; Min, E.S.; Scully, S.P. Prognostic Factors for Survival in Patients with Epithelioid Sarcoma: 441 Cases from the SEER Database. Clin. Orthop. Relat. Res. 2009, 467, 2939. [Google Scholar] [CrossRef] [PubMed] [Green Version]





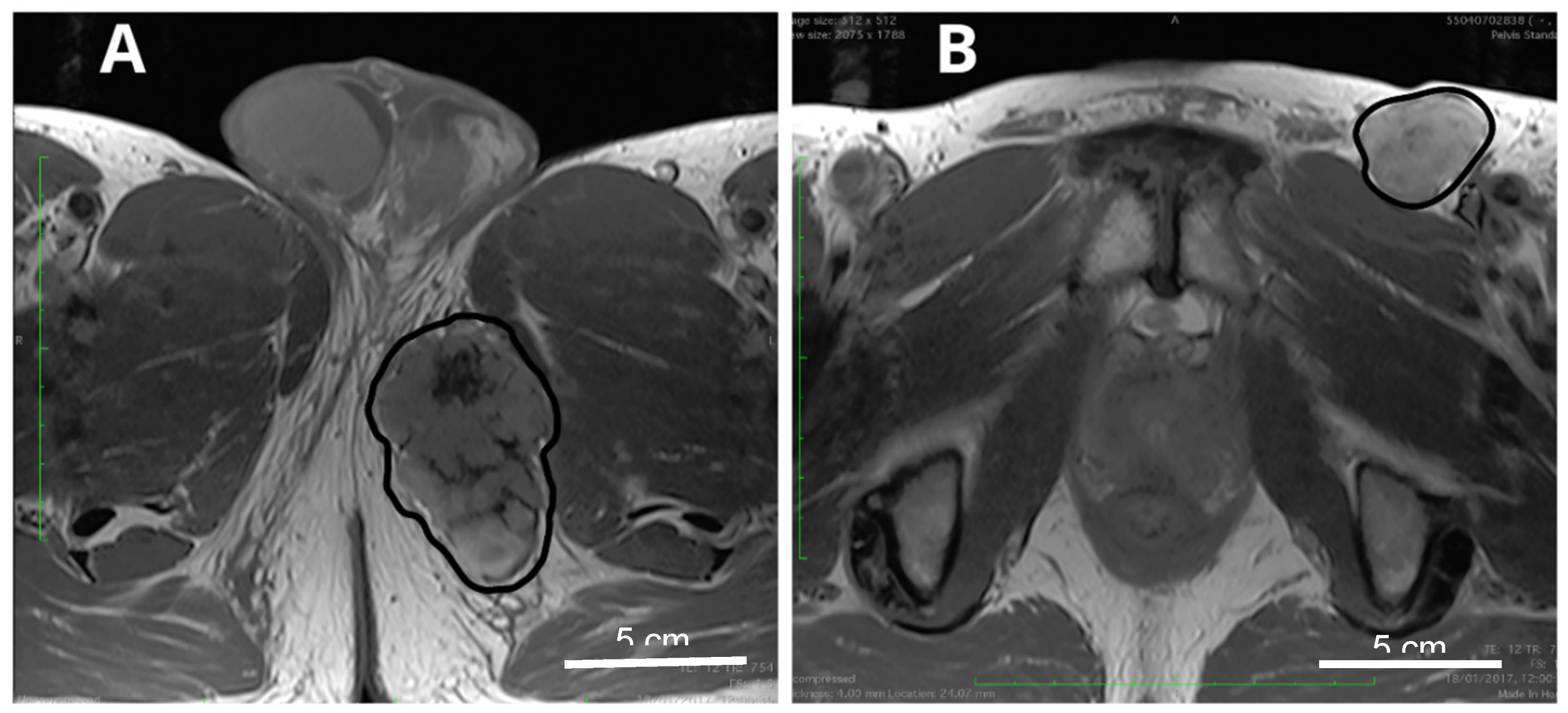{kind=link}
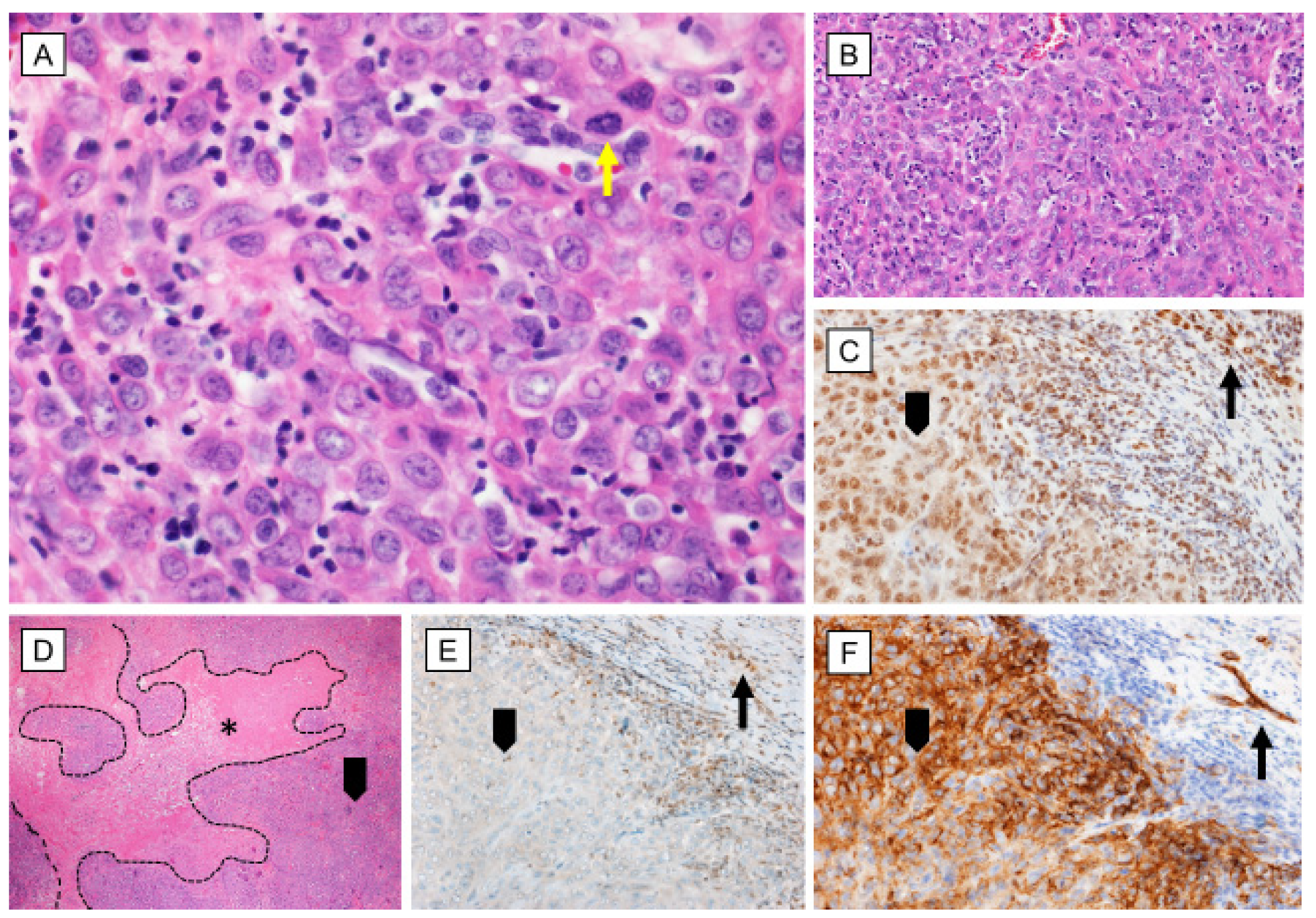{kind=link}
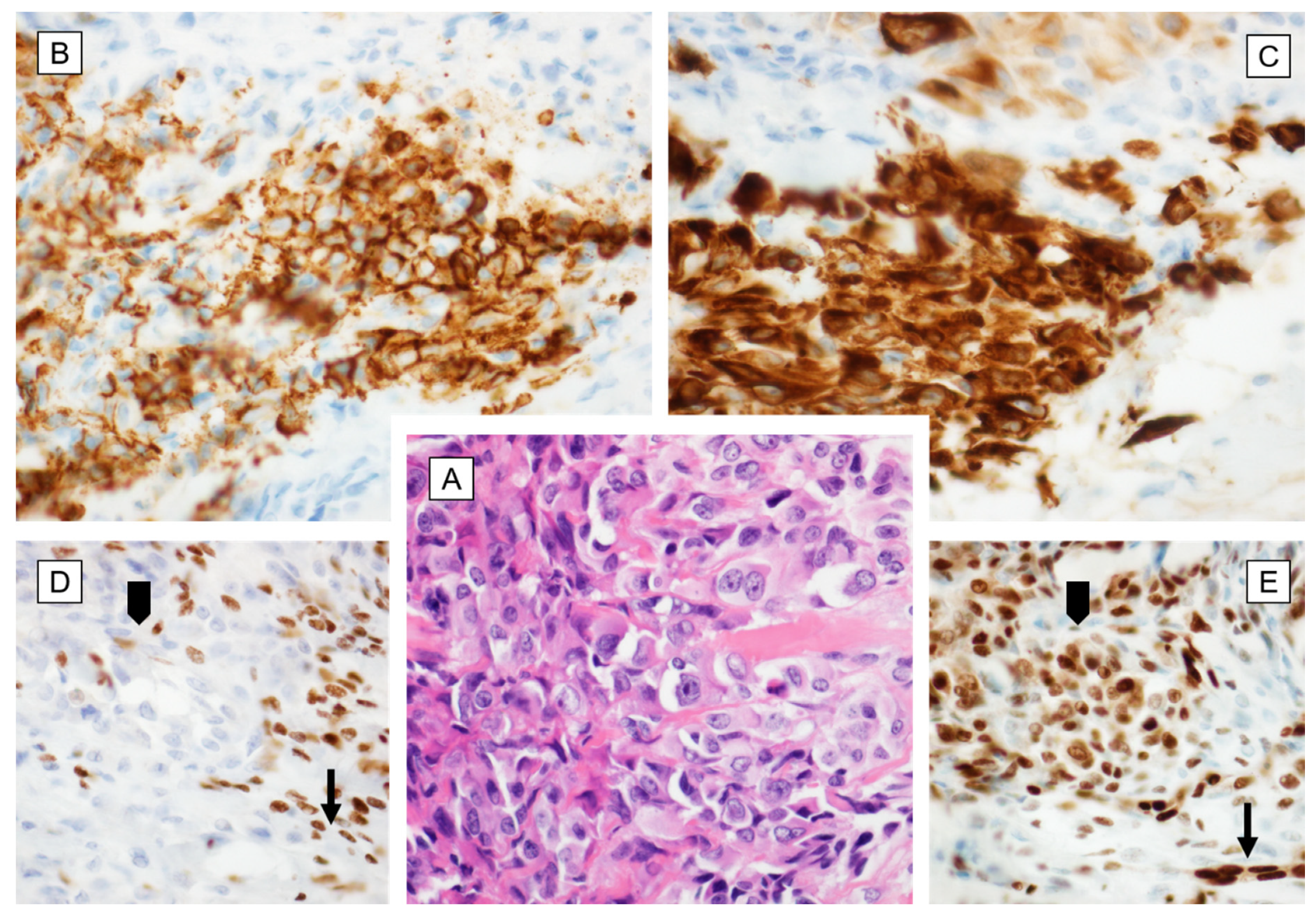{kind=link}
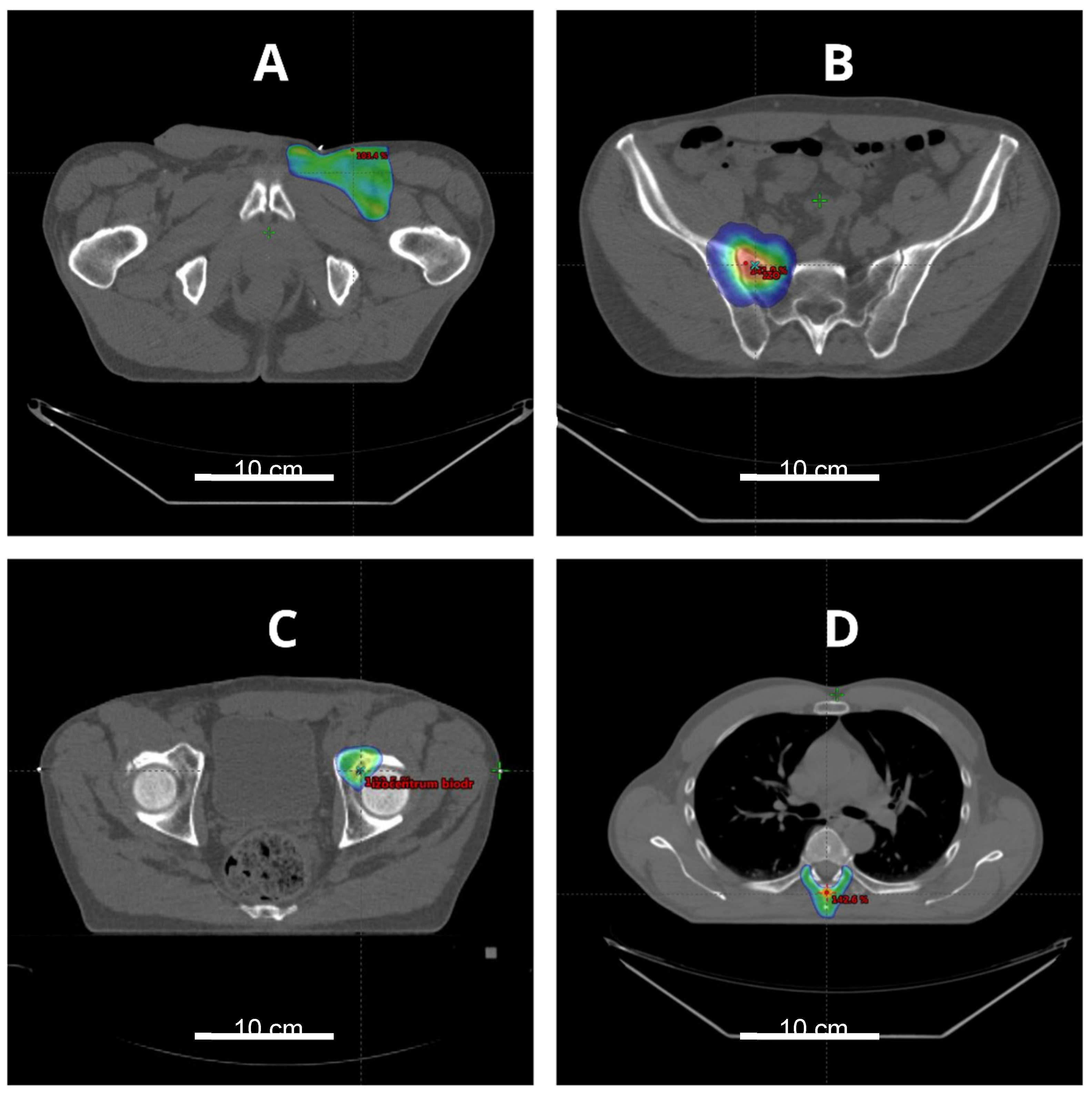{kind=link}
| Drugs | Dose | Frequency | ORR | Ref. |
|---|---|---|---|---|
| Doxorubicin | 75 mg/m2 | d. 1 every 3 weeks (bolus or 72h infusion) | NA 0% (0/14) * | [91] [96] |
| Doxorubicin Ifosfamide | 20 mg/m2 3 g/m2 | d. 1–3 every 3 weeks d. 1–3 every 3 weeks | NA | [91] |
| Doxorubicin Ifosfamide | 20 mg/m2 2 g/m2 | d. 1–3 every 3 weeks d. 1–3 every 3 weeks | NA | [31] |
| Doxorubicin Ifosfamide (perioperative, 3 cycles) | 20 mg/m2 1.5 mg/m2 | d. 1–3 every 3 weeks d. 1–4 every 3 weeks | NA | [92] |
| Doxorubicin Ifosfamide | 25 mg/m2 2,5 mg/m2 | d. 1–3 every 3 weeks d. 1–4 every 3 weeks | 12.5% (1/8) * | [96] |
| Anthracycline-based regimens | NA | NA | 19/85 (22%) # | [94] |
| Trabectedin | 1.5 mg/m2 | Every 3 weeks | NA | [91] |
| Ifosfamide Vincristine Doxorubicin (IVAD) | 3 g/m2 1.4 mg/m2 20 mg/m2 | d. 1–3 every 3 weeks d. 1 every 3 weeks d. 1–3 every 3 weeks | NA | [91] |
| Pazopanib | 800 mg | daily | 0% (0/18) # 100% (2/2) * 100% (2/2) ^ 100% (1/1) ^ | [94] [96] [99] [100] |
| Trabectedin | 1.5 mg/m2 | d. 1 every 3 weeks (24h infusion) | 33.3% (1/3) * | [96] |
| Gemcitabine Docetaxel | 900 mg/m2 100 mg/m2 | d. 1, 8 every 3 weeks d. 8 every 3 weeks | 58% (7/12) # | [95] |
| Gemcitabine-based | NA | NA | 27% (11/41) # | [94] |
| Vinorelbin | 17–30 mg/m2 | d. 1 every 2–4 weeks | 100% (1/1) ^ 100% (1/1) ^ | [97] [97,98] |
| Tazemetostat | 800 mg | bidaily | 15% (9/62) * | [50] |
| Dasatinib | 70 mg | bidaily | 28.6% (2/7) | [104] |
| Nivolumab | 3 mg/kg bw | every 2 weeks | 50% (1/2) # | [107] |
| Pembrolizumab | 2 mg/kg bw | every 3 weeks | 100% (1.1) * | [106] |
| Number | Drug | Status | Details | Additional Information |
|---|---|---|---|---|
| NCT04204941 | Doxorubicin + Tazemetostat/placebo | Recruiting | Tazemetostat: 400/600/800 mg BID Doxorubicin: 75 m2 d. 1 every 3 weeks for 6 cycles | Phase 1b/3 |
| NCT03009201 | Ribociclib + doxorubicin | Active, not recruiting | Ribociclib d. 1-6 400 mg and Doxorubicin 75 mg/m2 d. 10, every 3 weeks for 6 cycles, than ribocyclib daily | |
| NCT01532687 | Gemcitabine + pazopanib/placebo | Active, not recruiting | NA | |
| NCT01154452 | Vismodegib + Gamma–Secretase/Notch Signaling Pathway Inhibitor RO4929097 or placebo | Completed | Vismodegib 150 mg QD and RO4929097 15 mg QD | No specific results for epithelioid sarcoma available |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnecka, A.M.; Sobczuk, P.; Kostrzanowski, M.; Spalek, M.; Chojnacka, M.; Szumera-Cieckiewicz, A.; Rutkowski, P. Epithelioid Sarcoma—From Genetics to Clinical Practice. Cancers 2020, 12, 2112. https://doi.org/10.3390/cancers12082112
Czarnecka AM, Sobczuk P, Kostrzanowski M, Spalek M, Chojnacka M, Szumera-Cieckiewicz A, Rutkowski P. Epithelioid Sarcoma—From Genetics to Clinical Practice. Cancers. 2020; 12(8):2112. https://doi.org/10.3390/cancers12082112
Chicago/Turabian StyleCzarnecka, Anna M., Pawel Sobczuk, Michal Kostrzanowski, Mateusz Spalek, Marzanna Chojnacka, Anna Szumera-Cieckiewicz, and Piotr Rutkowski. 2020. "Epithelioid Sarcoma—From Genetics to Clinical Practice" Cancers 12, no. 8: 2112. https://doi.org/10.3390/cancers12082112