Upregulation of Protein Synthesis and Proteasome Degradation Confers Sensitivity to Proteasome Inhibitor Bortezomib in Myc-Atypical Teratoid/Rhabdoid Tumors

 , , , , , , , , and add
Show full author list
, , , , , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
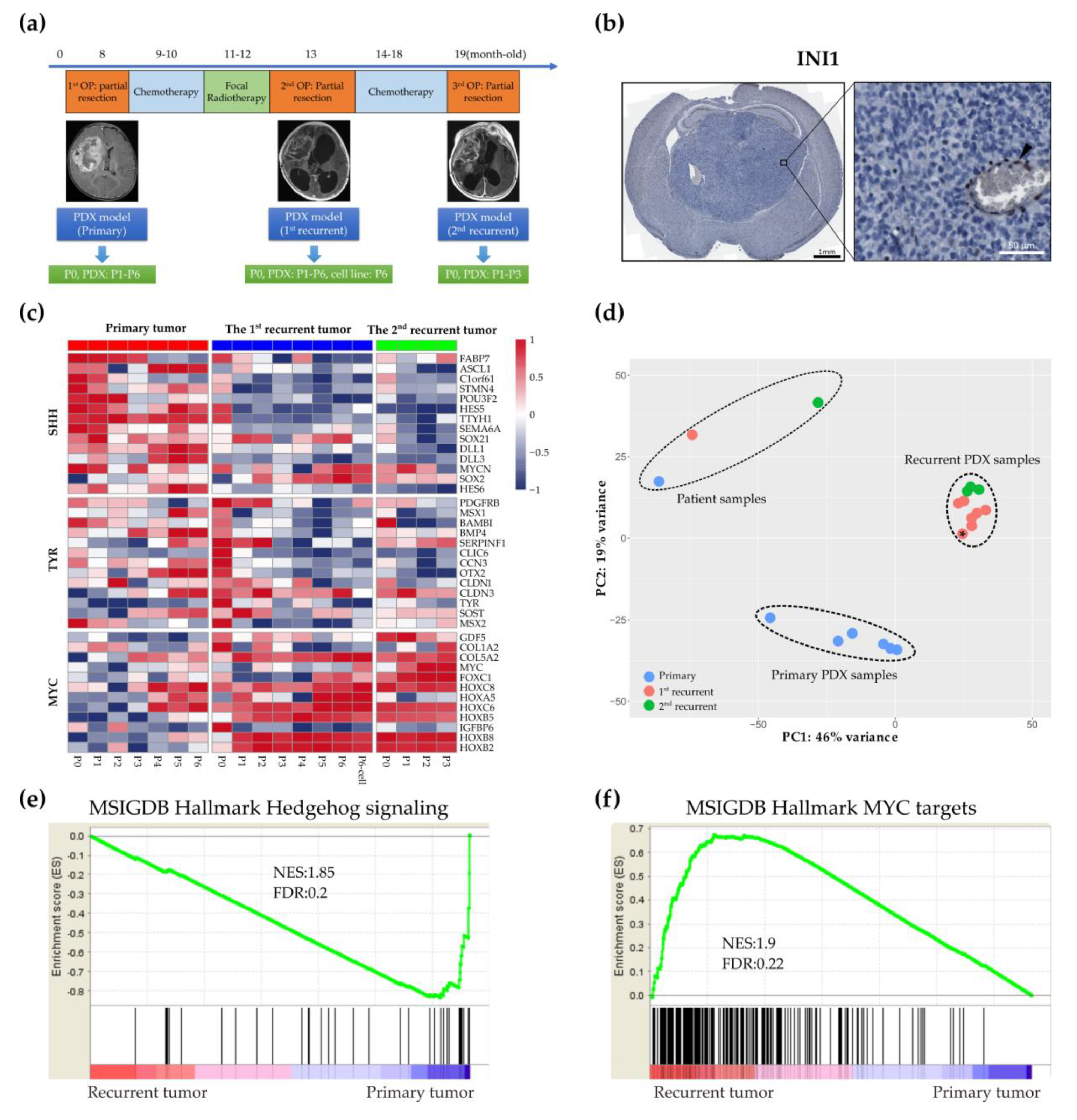
2.1. Establishing a Matched Model for the Primary and Recurrent Atypical Teratoid Rhabdoid Tumors
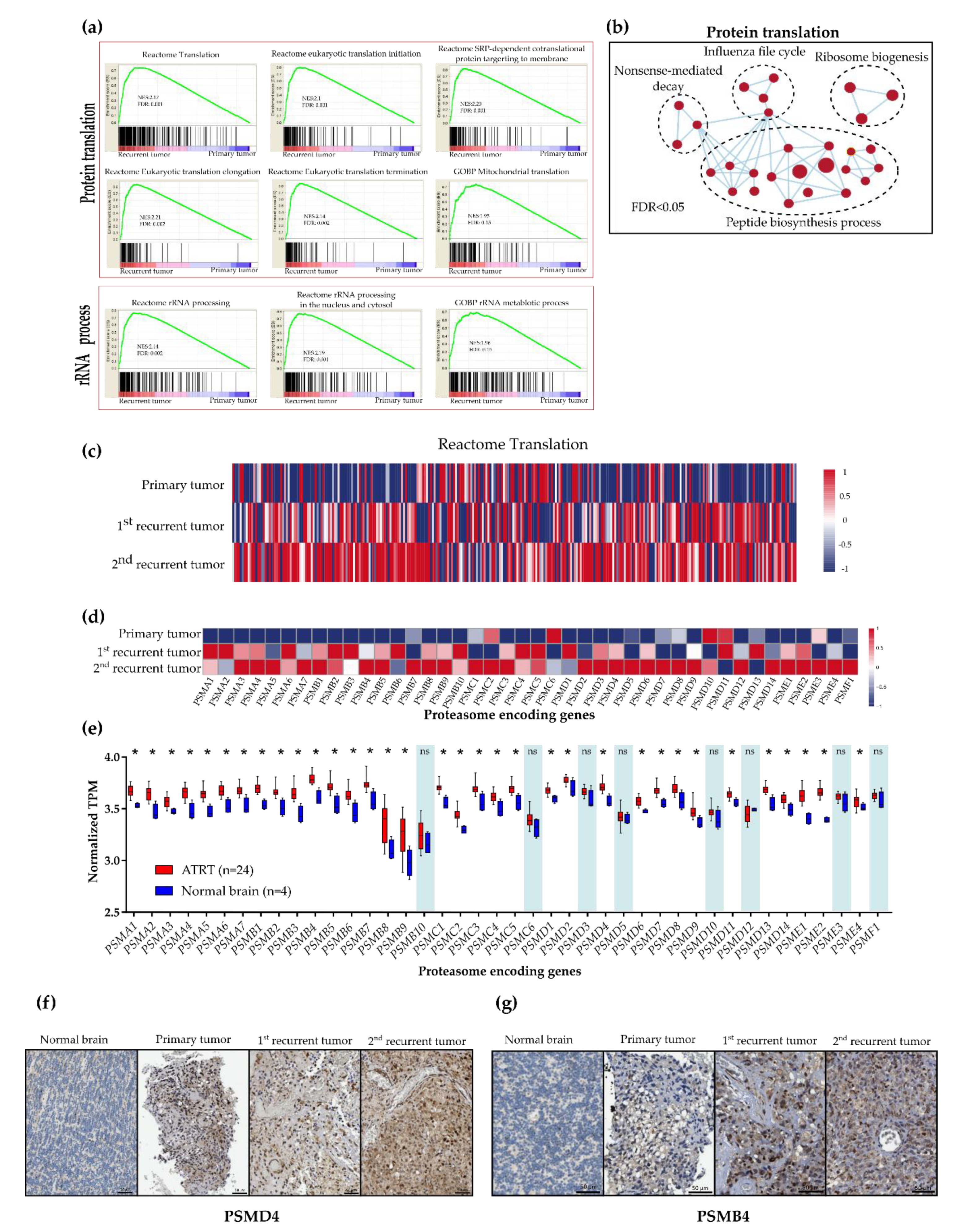
2.2. Upregulation of Protein Synthesis and Proteasome Degradation in Recurrent ATRT
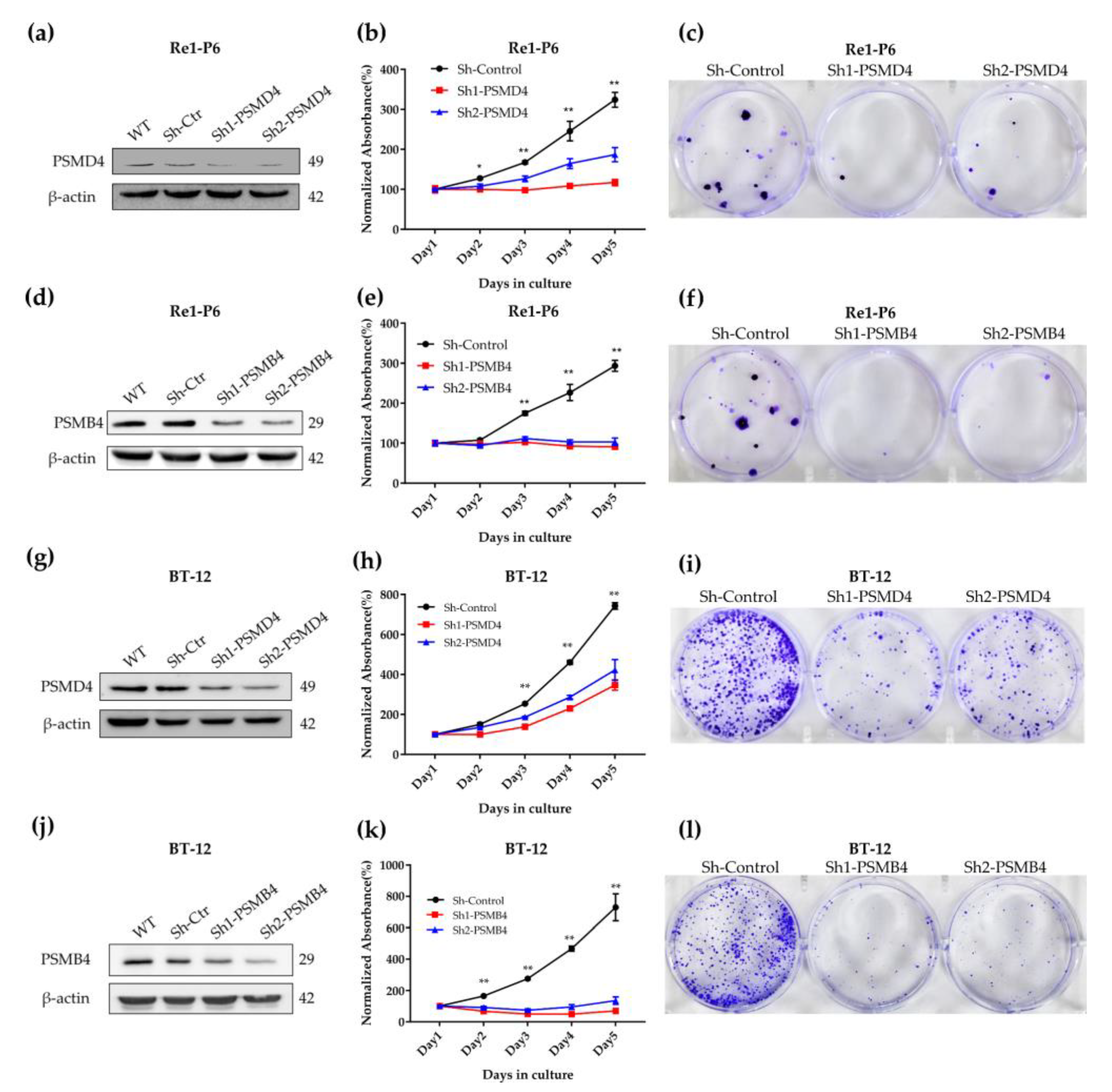
2.3. Importance of PSMD4 and PSMB4 in ATRT Cell Survival
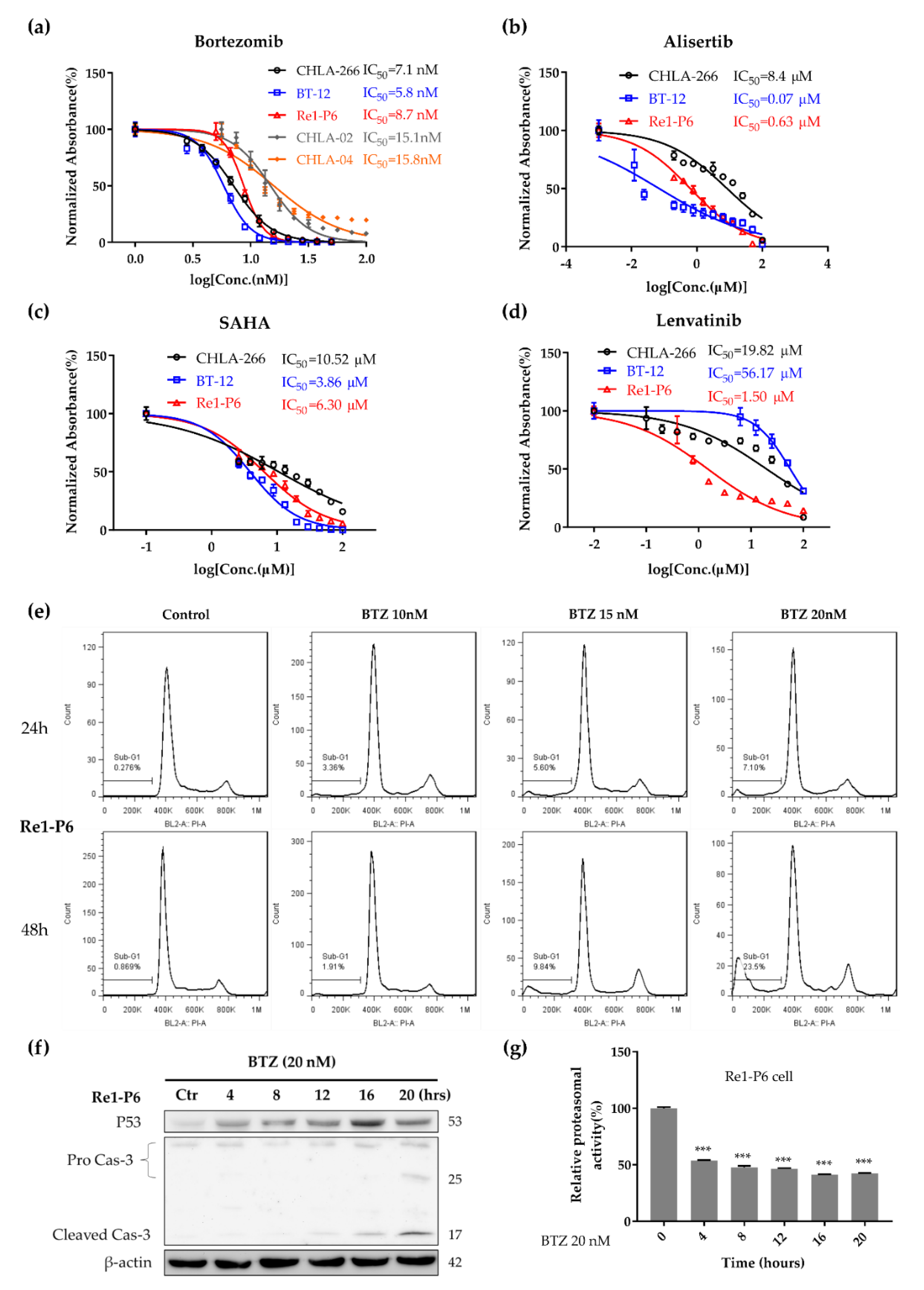
2.4. Proteasome Inhibitor BTZ Inhibited Tumor Growth and Induced Apoptosis In Vitro
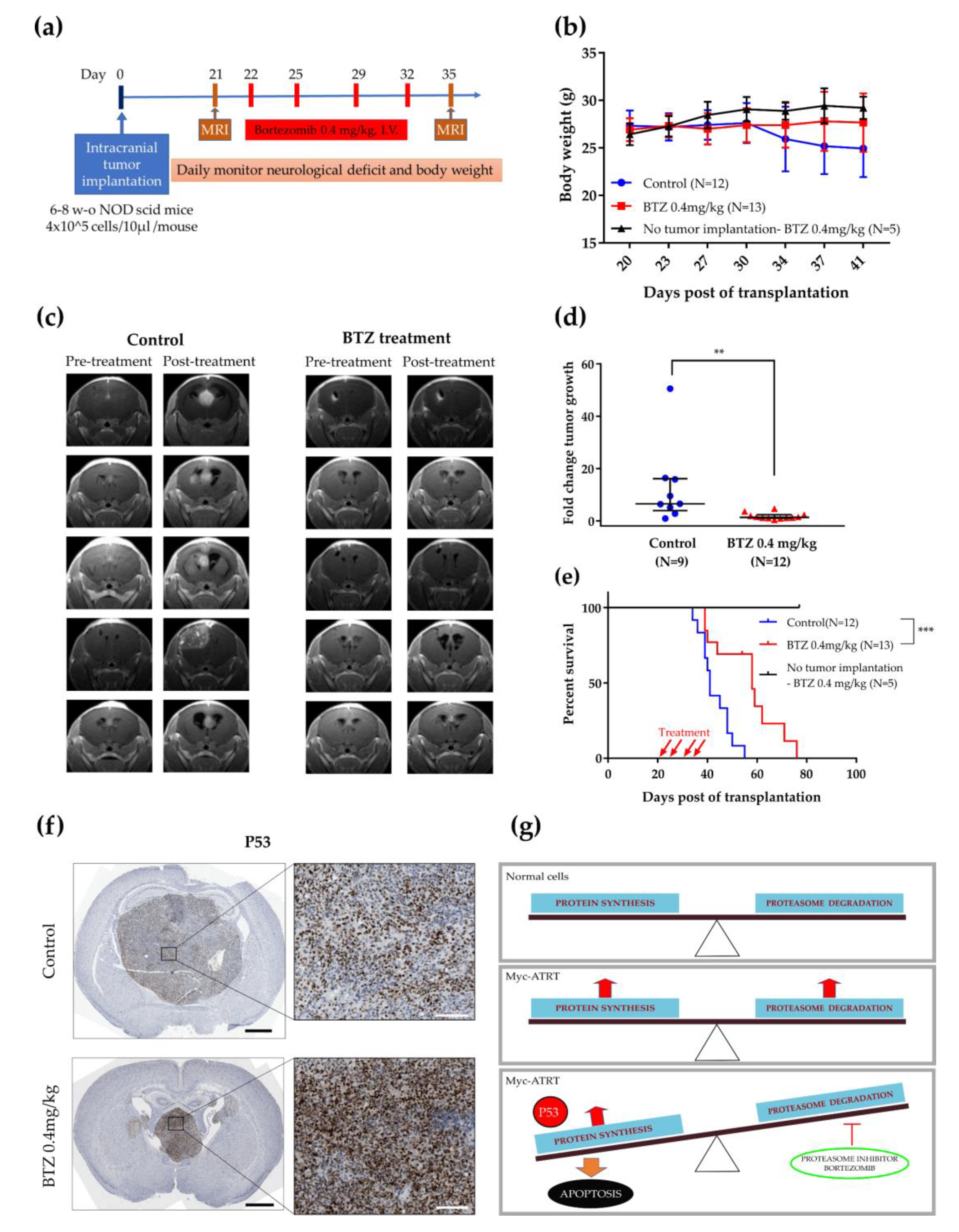
2.5. BTZ Inhibited Tumor Growth and Prolonged Survival Time in the Orthotopic Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Cells Cultures
4.2. Cell Growth Inhibition Assay
4.3. Colony Formation Assay
4.4. Apoptosis Assay
4.5. Immunohistochemistry
4.6. Western Blots
4.7. Immunoprecipitation
4.8. Proteasome Activity Assay
4.9. Lentiviral Transduction
4.10. Establishing PDX and Orthotopic Human ATRT Xenograft Models
4.11. RNA-Seq Analysis, Gene Set Enrichment Analysis and Whole-Exome Sequencing Analysis
4.12. Sanger Sequencing
4.13. Data Availability
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; de Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s lemonade stand foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro-Oncology 2015, 16 (Suppl. 10), x1–x36. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.A.; Ho, B.; Huang, A. Atypical teratoid rhabdoid tumour: From tumours to therapies. J. Korean Neurosurg. Soc. 2018, 61, 302–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807. [Google Scholar] [CrossRef]
- Wojcik, C. Regulation of apoptosis by the ubiquitin and proteasome pathway. J. Cell. Mol. Med. 2002, 6, 25–48. [Google Scholar] [CrossRef]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [Green Version]
- San Miguel, J.F.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; Samoilova, O.S.; et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N. Engl. J. Med. 2008, 359, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Robak, T.; Huang, H.; Jin, J.; Zhu, J.; Liu, T.; Samoilova, O.; Pylypenko, H.; Verhoef, G.; Siritanaratkul, N.; Osmanov, E.; et al. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N. Engl. J. Med. 2015, 372, 944–953. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J., Jr.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Cenci, S.; Oliva, L.; Cerruti, F.; Milan, E.; Bianchi, G.; Raule, M.; Mezghrani, A.; Pasqualetto, E.; Sitia, R.; Cascio, P. Pivotal Advance: Protein synthesis modulates responsiveness of differentiating and malignant plasma cells to proteasome inhibitors. J. Leukoc. Biol. 2012, 92, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Torchia, J.; Golbourn, B.; Feng, S.; Ho, K.C.; Sin-Chan, P.; Vasiljevic, A.; Norman, J.D.; Guilhamon, P.; Garzia, L.; Agamez, N.R.; et al. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell 2016, 30, 891–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.W.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 2016, 29, 379–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggero, D. The role of Myc-induced protein synthesis in cancer. Cancer Res. 2009, 69, 8839–8843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Birks, D.K.; Donson, A.M.; Patel, P.R.; Sufit, A.; Algar, E.M.; Dunham, C.; Kleinschmidt-DeMasters, B.K.; Handler, M.H.; Vibhakar, R.; Foreman, N.K. Pediatric rhabdoid tumors of kidney and brain show many differences in gene expression but share dysregulation of cell cycle and epigenetic effector genes. Pediatr. Blood Cancer 2013, 60, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Birks, D.K.; Donson, A.M.; Patel, P.R.; Dunham, C.; Muscat, A.; Algar, E.M.; Ashley, D.M.; Kleinschmidt-Demasters, B.K.; Vibhakar, R.; Handler, M.H.; et al. High expression of BMP pathway genes distinguishes a subset of atypical teratoid/rhabdoid tumors associated with shorter survival. Neuro-Oncology 2011, 13, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.-Y.; Richer, W.; Fréneaux, P.; Chauvin, C.; Lucchesi, C.; Guillemot, D.; Grison, C.; Lequin, D.; Pierron, G.; Masliah-Planchon, J.; et al. The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat. Commun. 2016, 7, 10421. [Google Scholar] [CrossRef] [Green Version]
- Roth, R.B.; Hevezi, P.; Lee, J.; Willhite, D.; Lechner, S.M.; Foster, A.C.; Zlotnik, A. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics 2006, 7, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a cancer dependency map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knipstein, J.A.; Birks, D.K.; Donson, A.M.; Alimova, I.; Foreman, N.K.; Vibhakar, R. Histone deacetylase inhibition decreases proliferation and potentiates the effect of ionizing radiation in atypical teratoid/rhabdoid tumor cells. Neuro-Oncology 2012, 14, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar] [PubMed]
- Hideshima, T.; Mitsiades, C.; Akiyama, M.; Hayashi, T.; Chauhan, D.; Richardson, P.; Schlossman, R.; Podar, K.; Munshi, N.C.; Mitsiades, N.; et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood 2003, 101, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Weissmiller, A.M.; Wang, J.; Lorey, S.L.; Howard, G.C.; Martinez, E.; Liu, Q.; Tansey, W.P. Inhibition of MYC by the SMARCB1 tumor suppressor. Nat. Commun. 2019, 10, 2014. [Google Scholar] [CrossRef] [Green Version]
- Carugo, A.; Minelli, R.; Sapio, L.; Soeung, M.; Carbone, F.; Robinson, F.S.; Tepper, J.; Chen, Z.; Lovisa, S.; Svelto, M.; et al. p53 Is a master regulator of proteostasis in SMARCB1-deficient malignant rhabdoid tumors. Cancer Cell 2019, 35, 204–220. [Google Scholar] [CrossRef] [Green Version]
- Alimova, I.; Pierce, A.; Danis, E.; Donson, A.; Birks, D.K.; Griesinger, A.; Foreman, N.K.; Santi, M.; Soucek, L.; Venkataraman, S.; et al. Inhibition of MYC attenuates tumor cell self-renewal and promotes senescence in SMARCB1-deficient Group 2 atypical teratoid rhabdoid tumors to suppress tumor growth in vivo. Int. J. Cancer 2019, 144, 1983–1995. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of therapy with bortezomib in solid tumors: A review based on 32 clinical trials. Future Oncol. (Lond. Engl.) 2014, 10, 1795–1807. [Google Scholar] [CrossRef]
- Russo, S.M.; Tepper, J.E.; Baldwin, A.S., Jr.; Liu, R.; Adams, J.; Elliott, P.; Cusack, J.C., Jr. Enhancement of radiosensitivity by proteasome inhibition: Implications for a role of NF-kappaB. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 183–193. [Google Scholar] [CrossRef]
- Sunwoo, J.B.; Chen, Z.; Dong, G.; Yeh, N.; Crowl Bancroft, C.; Sausville, E.; Adams, J.; Elliott, P.; Van Waes, C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 1419–1428. [Google Scholar]
- Ling, Y.-H.; Liebes, L.; Jiang, J.-D.; Holland, J.F.; Elliott, P.J.; Adams, J.; Muggia, F.M.; Perez-Soler, R. Mechanisms of proteasome inhibitor PS-341-induced G(2)-M-phase arrest and apoptosis in human non-small cell lung cancer cell lines. Clin. Cancer Res. 2003, 9, 1145–1154. [Google Scholar]
- Williams, S.A.; McConkey, D.J. The proteasome inhibitor bortezomib stabilizes a novel active form of p53 in human LNCaP-Pro5 prostate cancer cells. Cancer Res. 2003, 63, 7338–7344. [Google Scholar] [PubMed]
- Mitsiades, C.S.; McMillin, D.; Kotoula, V.; Poulaki, V.; McMullan, C.; Negri, J.; Fanourakis, G.; Tseleni-Balafouta, S.; Ain, K.B.; Mitsiades, N. Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. J. Clin. Endocrinol. Metab. 2006, 91, 4013–4021. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Carroll, M.; Thomas-Tikhonenko, A. p53 status dictates responses of B lymphomas to monotherapy with proteasome inhibitors. Blood 2007, 109, 4936–4943. [Google Scholar] [CrossRef] [PubMed]
- Concannon, C.G.; Koehler, B.F.; Reimertz, C.; Murphy, B.M.; Bonner, C.; Thurow, N.; Ward, M.W.; Villunger, A.; Strasser, A.; Kogel, D.; et al. Apoptosis induced by proteasome inhibition in cancer cells: Predominant role of the p53/PUMA pathway. Oncogene 2007, 26, 1681–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Barker, N.; Hoon, S.; He, P.; Thakur, T.; Abdeen, S.R.; Maruthappan, P.; Ghadessy, F.J.; Lane, D.P. Bortezomib stabilizes and activates p53 in proliferative compartments of both normal and tumor tissues in vivo. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Dufour, C.; Beaugrand, A.; Le Deley, M.C.; Bourdeaut, F.; Andre, N.; Leblond, P.; Bertozzi, A.I.; Frappaz, D.; Rialland, X.; Fouyssac, F.; et al. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: A multicenter study. Cancer 2012, 118, 3812–3821. [Google Scholar] [CrossRef]
- Hong, A.L.; Tseng, Y.-Y.; Wala, J.A.; Kim, W.-J.; Kynnap, B.D.; Doshi, M.B.; Kugener, G.; Sandoval, G.J.; Howard, T.P.; Li, J.; et al. Renal medullary carcinomas depend upon SMARCB1 loss and are sensitive to proteasome inhibition. eLife 2019, 8, e44161. [Google Scholar] [CrossRef]
- Johann, P.; Chun, E.; Erkek, S.; Iskar, M.; Perlman, E.; Hasselblatt, M.; Pfister, S.M.; Marra, M.; Kool, M. ATRT-09. Whole genome and epigenome characterization links atrt-myc to a subgroup of renal rhabdoid tumors. Neuro-Oncology 2018, 20, i29. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, function, and antigen presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouette, A.; Trofimov, A.; Haberl, D.; Boucher, G.; Lavallée, V.-P.; D’Angelo, G.; Hébert, J.; Sauvageau, G.; Lemieux, S.; Perreault, C. Expression of immunoproteasome genes is regulated by cell-intrinsic and -extrinsic factors in human cancers. Sci. Rep. 2016, 6, 34019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lightcap, E.S.; McCormack, T.A.; Pien, C.S.; Chau, V.; Adams, J.; Elliott, P.J. Proteasome inhibition measurements: Clinical application. Clin. Chem. 2000, 46, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkers, C.R.; Verdoes, M.; Lichtman, E.; Fiebiger, E.; Kessler, B.M.; Anderson, K.C.; Ploegh, H.L.; Ovaa, H.; Galardy, P.J. Activity probe for in vivo profiling of the specificity of proteasome inhibitor bortezomib. Nat. Methods 2005, 2, 357–362. [Google Scholar] [CrossRef]
- Li, X.; Kusmierczyk, A.R.; Wong, P.; Emili, A.; Hochstrasser, M. beta-Subunit appendages promote 20S proteasome assembly by overcoming an Ump1-dependent checkpoint. EMBO J. 2007, 26, 2339–2349. [Google Scholar] [CrossRef] [Green Version]
- D’Cunja, J.; Shalaby, T.; Rivera, P.; von Büren, A.; Patti, R.; Heppner, F.L.; Arcaro, A.; Rorke-Adams, L.B.; Phillips, P.C.; Grotzer, M.A. Antisense treatment of IGF-IR induces apoptosis and enhances chemosensitivity in central nervous system atypical teratoid/rhabdoid tumours cells. Eur. J. Cancer (Oxf. Engl. 1990) 2007, 43, 1581–1589. [Google Scholar] [CrossRef]
- Xu, J.; Erdreich-Epstein, A.; Gonzalez-Gomez, I.; Melendez, E.Y.; Smbatyan, G.; Moats, R.A.; Rosol, M.; Biegel, J.A.; Reynolds, C.P. Novel cell lines established from pediatric brain tumors. J. Neuro-Oncol. 2012, 107, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Margol, A.; Asgharzadeh, S.; Erdreich-Epstein, A. Pediatric brain tumor cell lines. J. Cell. Biochem. 2015, 116, 218–224. [Google Scholar] [CrossRef]
- Hsieh, T.-H.; Chien, C.-L.; Lee, Y.-H.; Lin, C.-I.; Hsieh, J.-Y.; Chao, M.-E.; Liu, D.-J.; Chu, S.-S.; Chen, W.; Lin, S.-C.; et al. Downregulation of SUN2, a novel tumor suppressor, mediates miR-221/222-induced malignancy in central nervous system embryonal tumors. Carcinogenesis 2014, 35, 2164–2174. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant review with the integrative genomics viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, H.M.; Wu, K.-S.; Sung, S.-Y.; Changou, C.A.; Hsieh, T.-H.; Liu, Y.-R.; Liu, Y.-L.; Tsai, M.-L.; Lee, H.-L.; Hsieh, K.L.-C.; et al. Upregulation of Protein Synthesis and Proteasome Degradation Confers Sensitivity to Proteasome Inhibitor Bortezomib in Myc-Atypical Teratoid/Rhabdoid Tumors. Cancers 2020, 12, 752. https://doi.org/10.3390/cancers12030752
Tran HM, Wu K-S, Sung S-Y, Changou CA, Hsieh T-H, Liu Y-R, Liu Y-L, Tsai M-L, Lee H-L, Hsieh KL-C, et al. Upregulation of Protein Synthesis and Proteasome Degradation Confers Sensitivity to Proteasome Inhibitor Bortezomib in Myc-Atypical Teratoid/Rhabdoid Tumors. Cancers. 2020; 12(3):752. https://doi.org/10.3390/cancers12030752
Chicago/Turabian StyleTran, Huy Minh, Kuo-Sheng Wu, Shian-Ying Sung, Chun Austin Changou, Tsung-Han Hsieh, Yun-Ru Liu, Yen-Lin Liu, Min-Lan Tsai, Hsin-Lun Lee, Kevin Li-Chun Hsieh, and et al. 2020. "Upregulation of Protein Synthesis and Proteasome Degradation Confers Sensitivity to Proteasome Inhibitor Bortezomib in Myc-Atypical Teratoid/Rhabdoid Tumors" Cancers 12, no. 3: 752. https://doi.org/10.3390/cancers12030752