Analysis of Cross-Association between mRNA Expression and RNAi Efficacy for Predictive Target Discovery in Colon Cancers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
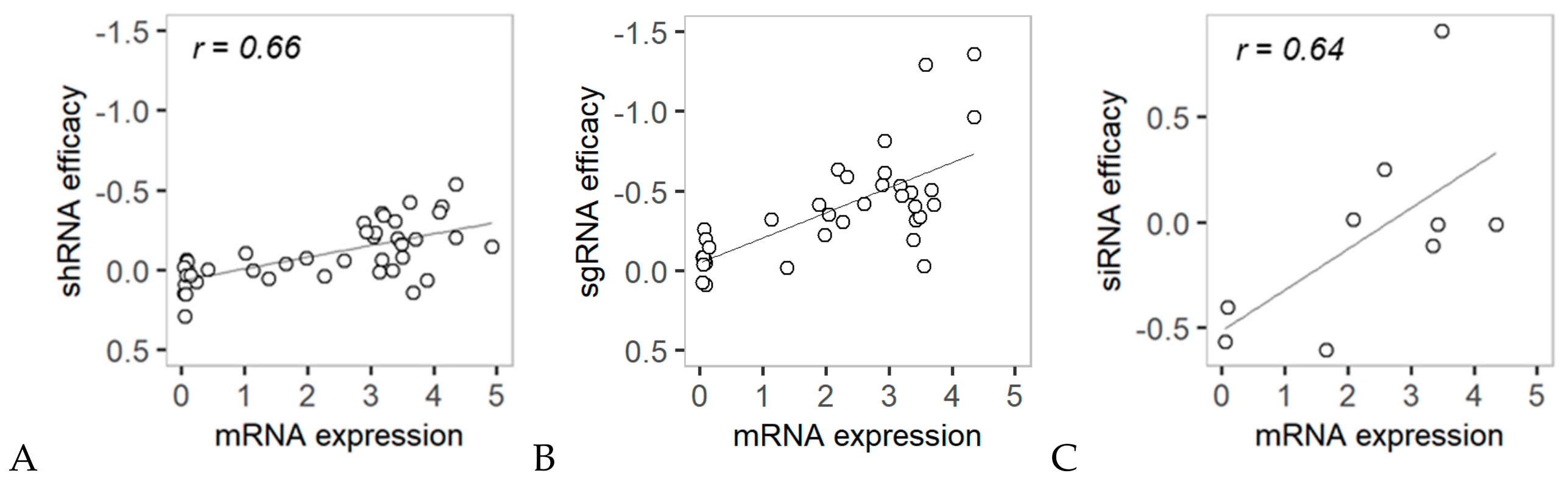
2.1. Overall Relationship between RNAi Efficacy and mRNA Expression Level
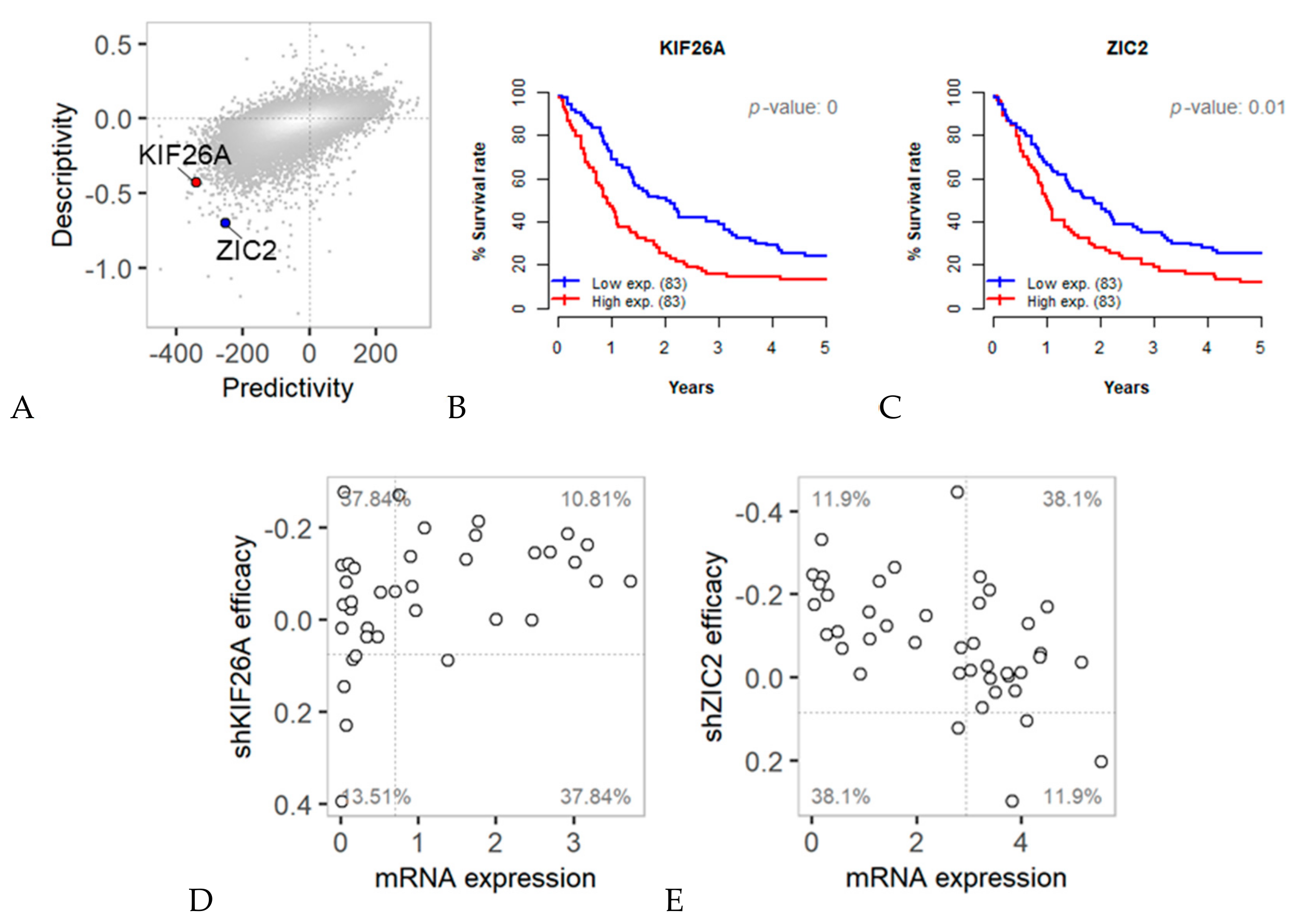
2.2. Predictivity and Descriptivity of Gene Expression for Anticancer RNAi Efficacy
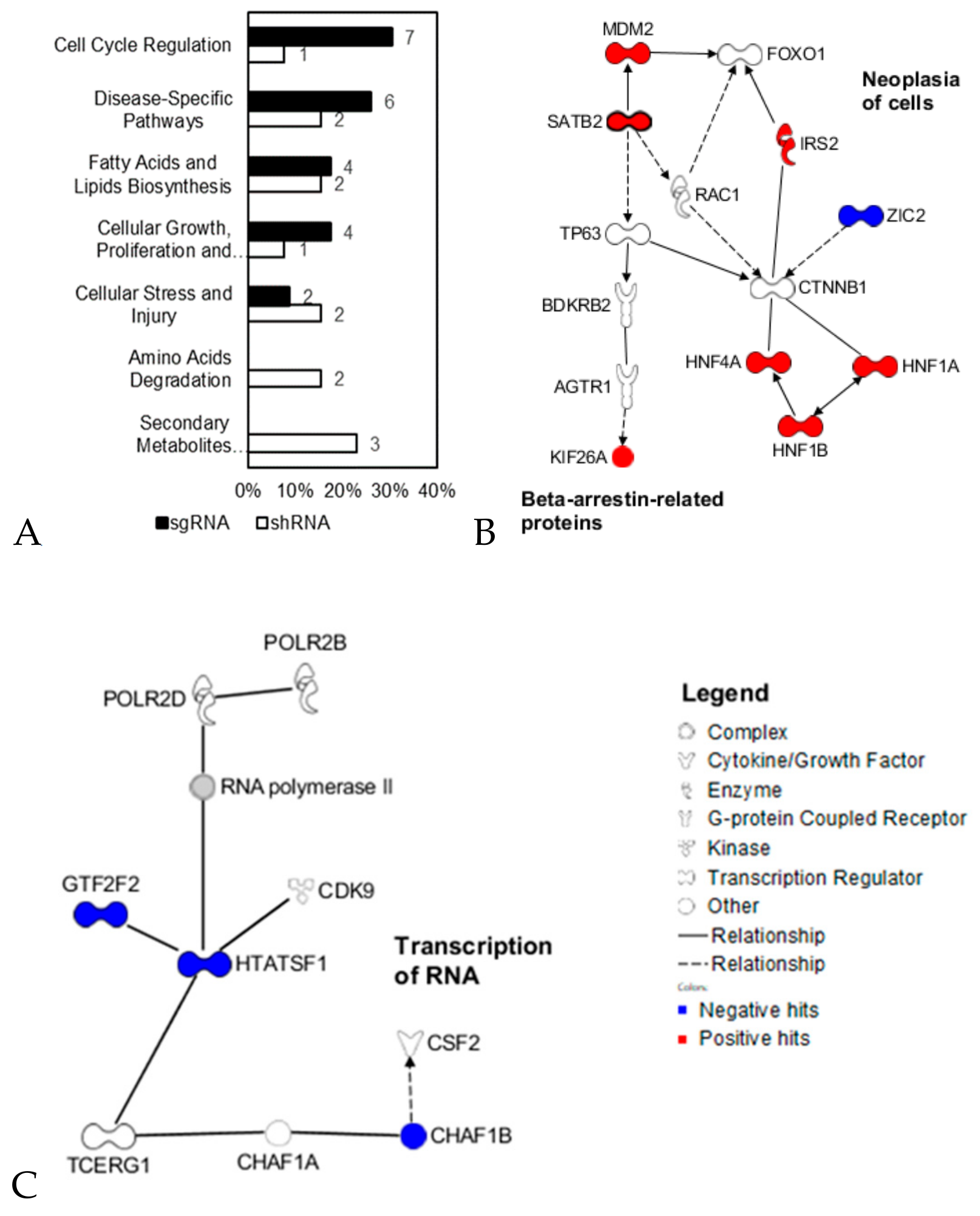
2.3. Functional Network Analysis of Predictive Target Genes
2.4. Survival Analysis of Predictive Anticancer Target Genes
3. Discussion
4. Materials and Methods
4.1. Acquisition of Data from Colon Cancer Cell Lines
4.2. Acquisition of Data from Patients with Colon Cancer
4.3. Bidirectional Analysis
4.4. Biological Functional Analysis and Network Construction
4.5. Cell Culture
4.6. Cell Viability and siRNA Transfection
4.7. Statistical Analyses for Bidirectional Analysis
4.8. Kaplan-Meier Survival Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Khleif, S.N.; Doroshow, J.H.; Hait, W.N. AACR-FDA-NCI Cancer Biomarkers Collaborative. AACR-FDA-NCI Cancer Biomarkers Collaborative consensus report: Advancing the use of biomarkers in cancer drug development. Clin. Cancer Res. 2010, 16, 3299–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, H.B. Predicting clinical outcomes using molecular biomarkers. Biomark. Cancer 2016, 8, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Mendiratta, P.; Grivas, P. Emerging biomarkers and targeted therapies in urothelial carcinoma. Ann. Transl. Med. 2018, 6, 250. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pasche, B.; Zhang, W.; Chen, K. Association of MUC16 mutation with tumor mutation load and outcomes in patients with gastric cancer. JAMA Oncol. 2018, 4, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, J.; Katta, A.; Masood, A.; Vudem, D.R.; Kancha, R.K. Emergence of ERBB2 mutation as a biomarker and an actionable target in solid cancers. Oncologist 2019, 24, e1303–e1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, P.K.; Li, J.; Jeong, K.J.; Shao, S.; Chen, H.; Tsang, Y.H.; Sengupta, S.; Wang, Z.; Bhavana, V.H.; Tran, R.; et al. Systematic functional annotation of somatic mutations in cancer. Cancer Cell 2018, 33, 450–462.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.; Song, M.; Kim, S.; Seo, Y.; Kim, Y.; Yoon, S. Differential regulation and synthetic lethality of exclusive RB1 and CDKN2A mutations in lung cancer. Int. J. Oncol. 2016, 48, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Lee, Y.; Je, S.; Chang, S.; Kim, N.; Jeong, E.; Yoon, S. Overexpression and selective anticancer efficacy of ENO3 in STK11 mutant lung cancers. Mol. Cells 2019, 42, 804–809. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Jiang, C.; Cao, S.; Li, N.; Jiang, L.; Sun, T. PD-1 and PD-L1 correlated gene expression profiles and their association with clinical outcomes of breast cancer. Cancer Cell Int. 2019, 19, 233. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the cancer cell line encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, J.M.; Ho, Z.V.; Kugener, G.; Dempster, J.M.; Montgomery, P.G.; Bryan, J.G.; Krill-Burger, J.M.; Green, T.M.; Vazquez, F.; Boehm, J.S.; et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, X.; Cheng, J.; Huang, Z.; Han, Z.; Helm, B.; Liu, X.; Zhang, J.; Wang, T.F.; Ni, D.; Huang, K. Correlation analysis of histopathology and proteogenomics data for breast cancer. Mol. Cell Proteom. 2019, 18, S37–S51. [Google Scholar] [CrossRef] [Green Version]
- Berg, K.C.G.; Eide, P.W.; Eilertsen, I.A.; Johannessen, B.; Bruun, J.; Danielsen, S.A.; Bjornslett, M.; Meza-Zepeda, L.A.; Eknaes, M.; Lind, G.E.; et al. Multi-omics of 34 colorectal cancer cell lines—A resource for biomedical studies. Mol. Cancer 2017, 16, 116. [Google Scholar] [CrossRef]
- Luo, Z.; Wang, W.; Li, F.; Songyang, Z.; Feng, X.; Xin, C.; Dai, Z.; Xiong, Y. Pan-cancer analysis identifies telomerase-associated signatures and cancer subtypes. Mol. Cancer 2019, 18, 106. [Google Scholar] [CrossRef] [Green Version]
- Ayiomamitis, G.D.; Notas, G.; Vasilakaki, T.; Tsavari, A.; Vederaki, S.; Theodosopoulos, T.; Kouroumalis, E.; Zaravinos, A. Understanding the interplay between COX-2 and hTERT in colorectal cancer using a multi-omics analysis. Cancers 2019, 11, 1536. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.A. Should Pearson’s correlation coefficient be avoided? Ophthalmic Physiol. Opt. 2019, 39, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Hardin, J.; Mitani, A.; Hicks, L.; VanKoten, B. A robust measure of correlation between two genes on a microarray. BMC Bioinform. 2007, 8, 220. [Google Scholar] [CrossRef] [Green Version]
- Peretz, L.; Besser, E.; Hajbi, R.; Casden, N.; Ziv, D.; Kronenberg, N.; Gigi, L.B.; Sweetat, S.; Khawaled, S.; Aqeilan, R.; et al. Combined shRNA over CRISPR/cas9 as a methodology to detect off-target effects and a potential compensatory mechanism. Sci. Rep. 2018, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Morgens, D.W.; Deans, R.M.; Li, A.; Bassik, M.C. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 2016, 34, 634–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evers, B.; Jastrzebski, K.; Heijmans, J.P.; Grernrum, W.; Beijersbergen, R.L.; Bernards, R. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 2016, 34, 631–633. [Google Scholar] [CrossRef]
- Wang, J.; He, C.; Gao, P.; Wang, S.; Lv, R.; Zhou, H.; Zhou, Q.; Zhang, K.; Sun, J.; Fan, C.; et al. HNF1B-mediated repression of SLUG is suppressed by EZH2 in aggressive prostate cancer. Oncogene 2020, 39, 1335–1346. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Liu, L.; Hamada, T.; Nowak, J.A.; Giannakis, M.; Ma, Y.; Song, M.; Nevo, D.; Kosumi, K.; Gu, M.; et al. Night-shift work duration and risk of colorectal cancer according to IRS1 and IRS2 expression. Cancer Epidemiol. Biomark. Prev. 2020, 29, 133–140. [Google Scholar] [CrossRef]
- Wang, X.; Ji, P.; Zhang, Y.; LaComb, J.F.; Tian, X.; Li, E.; Williams, J.L. Aberrant DNA Methylation: Implications in racial health disparity. PLoS ONE 2016, 11, e0153125. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Olevian, D.; Miller, C.; Herbst, C.; Jayachandran, P.; Kozak, M.M.; Chang, D.T.; Pai, R.K. SATB2 and CDX2 are prognostic biomarkers in DNA mismatch repair protein deficient colon cancer. Mod. Pathol. 2019, 32, 1217–1231. [Google Scholar] [CrossRef]
- Depmap Portal. Available online: https://depmap.org/portal (accessed on 3 February 2020).
- GDC Data Portal. Available online: https://portal.gdc.cancer.gov (accessed on 3 February 2020).







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, E.; Lee, Y.; Kim, Y.; Lee, J.; Yoon, S. Analysis of Cross-Association between mRNA Expression and RNAi Efficacy for Predictive Target Discovery in Colon Cancers. Cancers 2020, 12, 3091. https://doi.org/10.3390/cancers12113091
Jeong E, Lee Y, Kim Y, Lee J, Yoon S. Analysis of Cross-Association between mRNA Expression and RNAi Efficacy for Predictive Target Discovery in Colon Cancers. Cancers. 2020; 12(11):3091. https://doi.org/10.3390/cancers12113091
Chicago/Turabian StyleJeong, Euna, Yejin Lee, Youngju Kim, Jieun Lee, and Sukjoon Yoon. 2020. "Analysis of Cross-Association between mRNA Expression and RNAi Efficacy for Predictive Target Discovery in Colon Cancers" Cancers 12, no. 11: 3091. https://doi.org/10.3390/cancers12113091