Inflammatory Breast Cancer: Clinical Implications of Genomic Alterations and Mutational Profiling

 , , ,
, , ,  , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
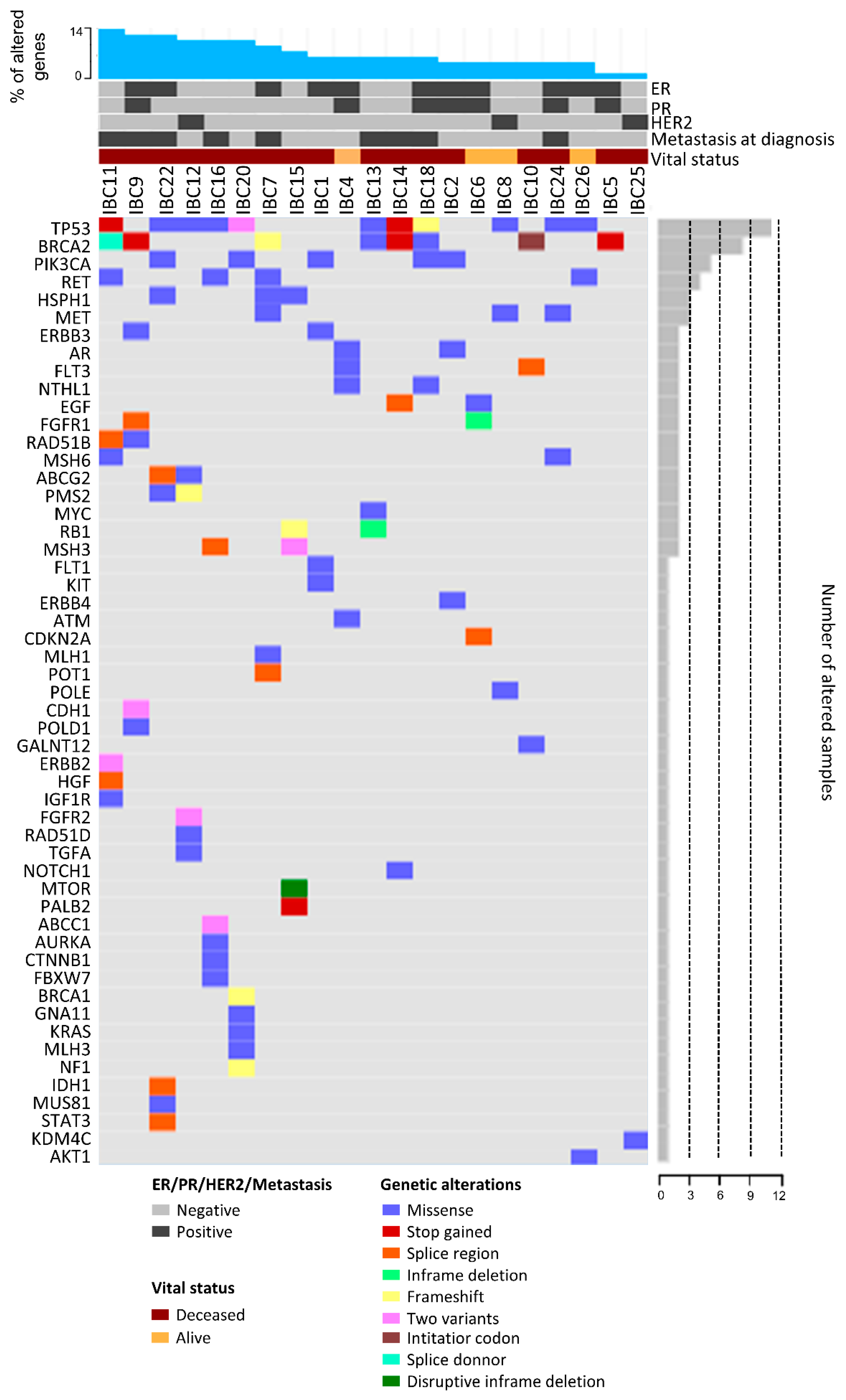
2.1. Clinical and Pathological Characteristics of IBC Patients
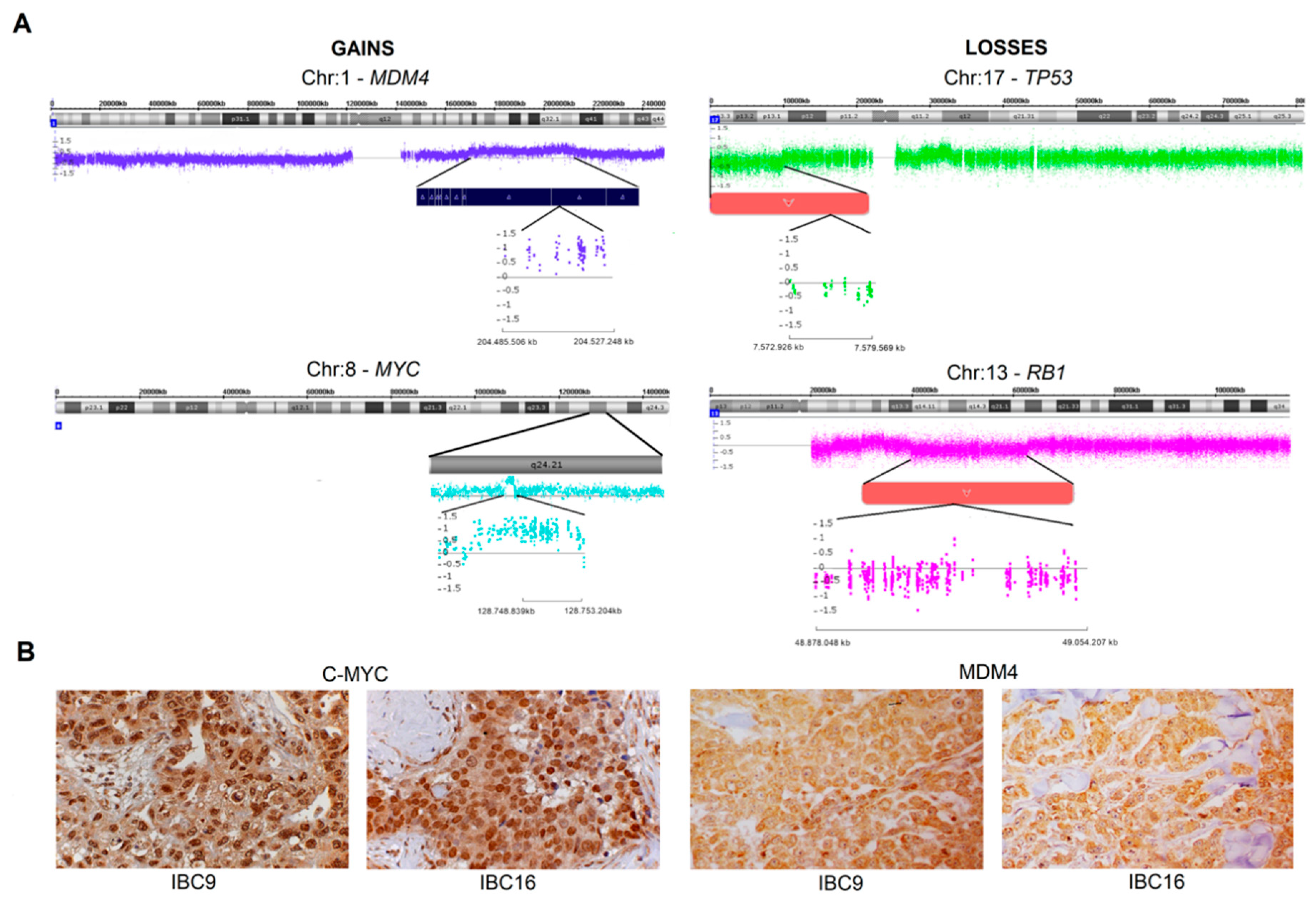
2.2. Genomic Profile-CNAs, cnLOH, and Chromothripsis (CTH)
2.3. MDM4 and C-MYC Proteins Expression
2.4. Genomic Instability (GII) Analysis and HRD-Related Genomic Scars Signatures
2.5. Mutational Profile
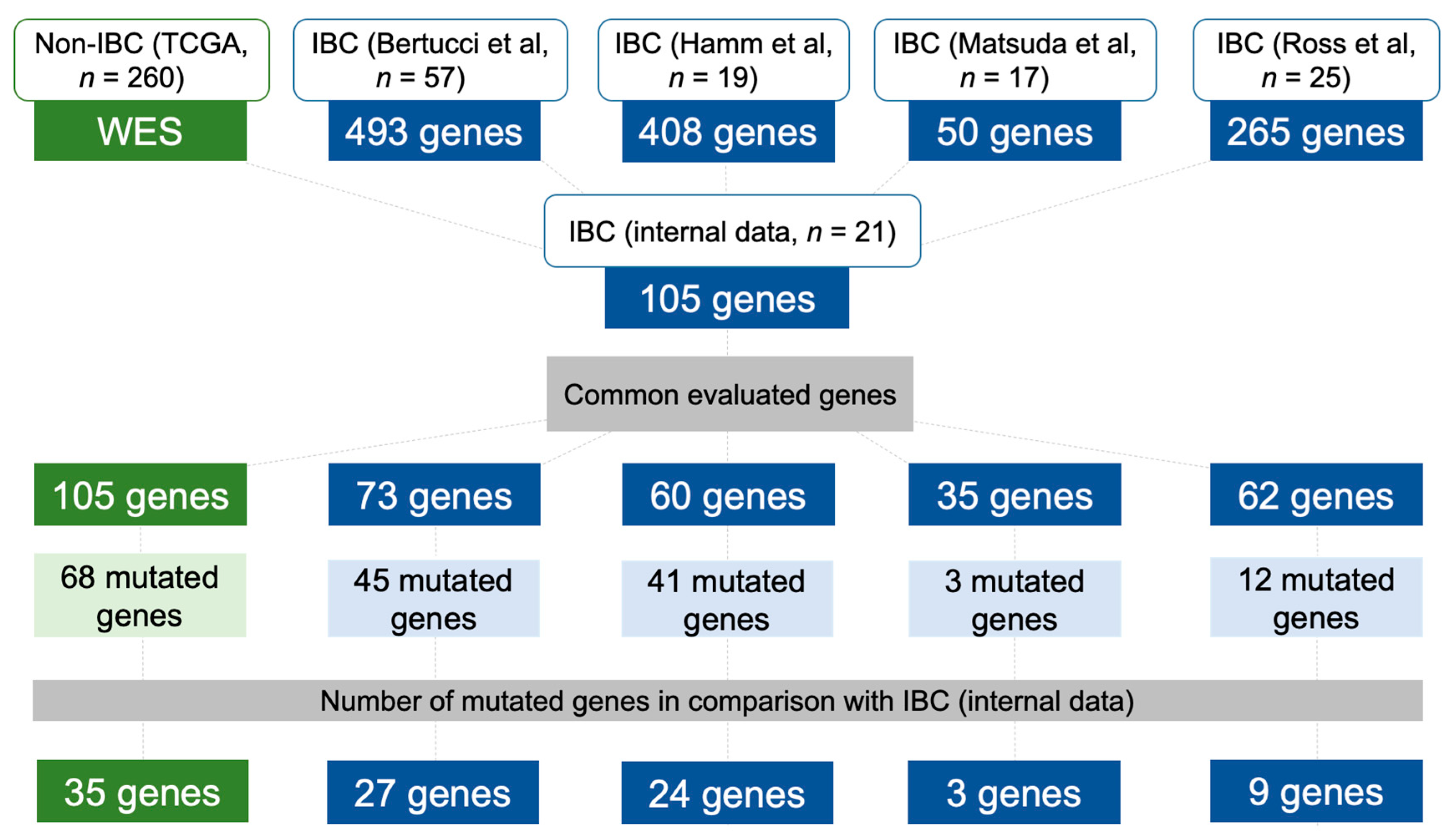
2.6. Data Comparison of the Mutational Profile in Non-IBC and IBC Cases
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. High-Resolution Chromosomal Microarray
4.3. Immunohistochemistry (IHC)
4.4. Target Enrichment-Next Generation Sequencing (tNGS) and Tumor Burden Assessment
4.5. Mutational Profile of Non-IBC and IBC Using Independent Validation Datasets
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hance, K.W.; Anderson, W.F.; Devessa, S.S.; Young, H.A.; Levine, P.H. Trends in inflammatory breast carcinoma incidence and survival: The surveillance, epidemiology, and end results program at the national cancer institute. J. Natl. Cancer Inst. 2005, 97, 966–975. [Google Scholar] [CrossRef]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef]
- Menta, A.; Fouad, T.M.; Lucci, A.; Le-Petross, H.; Stauder, M.C.; Woodward, W.A.; Ueno, N.T.; Lim, B. Inflammatory Breast Cancer: What to Know About This Unique, Aggressive Breast Cancer. Surg. Clin. North Am. 2018, 98, 787–800. [Google Scholar] [CrossRef]
- Dawood, S.; Merajver, S.D.; Viens, P.; Vermeulen, P.B.; Swain, S.M.; Buchholz, T.A.; Dirix, L.Y.; Levine, P.H.; Lucci, A.; Krishnamurthy, S.; et al. International expert panel on inflammatory breast cancer: Consensus statement for standardized diagnosis and treatment. Ann. Oncol. 2011, 22, 515–523. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Vermeulen, P.; Van Dam, P.; Dirix, L.; Birnbaum, D.; Viens, P.; Van Laere, S. Genomic profiling of inflammatory breast cancer: A review. Breast 2014, 23, 538–545. [Google Scholar] [CrossRef]
- Lehman, H.L.; Dashner, E.J.; Lucey, M.; Vermeulen, P.; Dirix, L.; Van Laere, S.; van Golen, K.L. Modeling and characterization of inflammatory breast cancer emboli grown in vitro. Int. J. Cancer 2013, 132, 2283–2294. [Google Scholar] [CrossRef]
- Schlichting, J.A.; Soliman, A.S.; Schairer, C.; Schottenfeld, D.; Merajver, S.D. Inflammatory and non-inflammatory breast cancer survival by socioeconomic position in the Surveillance, Epidemiology, and End Results database, 1990–2008. Breast Cancer Res. Treat. 2012, 134, 1257–1268. [Google Scholar] [CrossRef]
- Fouad, T.M.; Barrera, A.; Reuben, J.M.; Lucci, A.; Woodward, W.A.; Stauder, M.C.; Lim, B.; DeSnyder, S.M.; Arun, B.; Gildy, B.; et al. Inflammatory breast cancer: A proposed conceptual shift in the UICC-AJCC TNM staging system. Lancet Oncol. 2017, 18, e228–e232. [Google Scholar] [CrossRef]
- Liang, X.; Vacher, S.; Boulai, A.; Bernard, V.; Baulande, S.; Bohec, M.; Bièche, I.; Lerebours, F.; Callens, C. Targeted next-generation sequencing identifies clinically relevant somatic mutations in a large cohort of inflammatory breast cancer. Breast Cancer Res. 2018, 20, 88. [Google Scholar] [CrossRef]
- Bertucci, F.; Rypens, C.; Finetti, P.; Guille, A.; Adélaïde, J.; Monneur, A.; Carbuccia, N.; Garnier, S.; Dirix, P.; Gonçalves, A.; et al. NOTCH and DNA repair pathways are more frequently targeted by genomic alterations in inflammatory than in non-inflammatory breast cancers. Mol. Oncol. 2020, 14, 504–519. [Google Scholar] [CrossRef] [Green Version]
- Van Laere, S.J.; Ueno, N.T.; Finetti, P.; Vermeulen, P.; Lucci, A.; Robertson, F.M.; Marsan, M.; Iwamoto, T.; Krishnamurthy, S.; Masuda, H.; et al. Uncovering the molecular secrets of inflammatory breast cancer biology: An integrated analysis of three distinct affymetrix gene expression datasets. Clin. Cancer Res. 2013, 19, 4685–4696. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, F.; Ueno, N.T.; Finetti, P.; Vermeulen, P.; Lucci, A.; Robertson, F.M.; Marsan, M.; Iwamoto, T.; Krishnamurthy, S.; Masuda, H.; et al. Gene expression profiles of inflammatory breast cancer: Correlation with response to neoadjuvant chemotherapy and metastasis-free survival. Ann. Oncol. 2014, 25, 358–365. [Google Scholar] [CrossRef]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Iwamoto, T.; Brewer, T.; Pusztai, L.; Kai, K.; Kogawa, T.; Finetti, P.; Birnbaum, D.; et al. Comparison of molecular subtype distribution in triple-negative inflammatory and non-inflammatory breast cancers. Breast Cancer Res. 2013, 15, R112. [Google Scholar] [CrossRef] [Green Version]
- Ahomadegbe, J.C.; Tourpin, S.; Kaghad, M.; Zelek, L.; Vayssade, M.; Mathieu, M.C.; Rochard, F.; Spielmann, M.; Tursz, T.; Caput, D.; et al. Loss of heterozygosity, allele silencing and decreased expression of p73 gene in breast cancers: Prevalence of alterations in inflammatory breast cancers. Oncogene 2000, 19, 5413–5418. [Google Scholar] [CrossRef] [Green Version]
- Lerebours, F.; Bertheau, P.; Bieche, I.; Driouch, K.; De The, H.; Hacene, K.; Espie, M.; Marty, M.; Lidereau, R. Evidence of chromosome regions and gene involvement in inflammatory breast cancer. Int. J. Cancer. 2002, 102, 618–622. [Google Scholar] [CrossRef]
- Bekhouche, I.; Finetti, P.; Adelaïde, J.; Ferrari, A.; Tarpin, C.; Charafe-Jauffret, E.; Charpin, C.; Houvenaeghel, G.; Jacquemier, J.; Bidaut, G.; et al. High-resolution comparative genomic hybridization of inflammatory breast cancer and identification of candidate genes. PLoS ONE 2011, 6, e16950. [Google Scholar] [CrossRef] [Green Version]
- Woodward, W.A.; Krishnamurthy, S.; Yamauchi, H.; El-Zein, R.; Ogura, D.; Kitadai, E.; Niwa, S.; Cristofanilli, M.; Vermeulen, P.; Dirix, L.; et al. Genomic and expression analysis of microdissected inflammatory breast cancer. Breast Cancer Res. Treat. 2013, 138, 761–772. [Google Scholar] [CrossRef]
- Ross, J.S.; Ali, S.M.; Wang, K.; Khaira, D.; Palma, N.A.; Chmielecki, J.; Palmer, G.A.; Morosini, D.; Elvin, J.A.; Fernandez, S.V.; et al. Comprehensive genomic profiling of inflammatory breast cancer cases reveals a high frequency of clinically relevant genomic alterations. Breast Cancer Res. Treat. 2015, 154, 155–162. [Google Scholar] [CrossRef]
- Matsuda, N.; Lim, B.; Wang, Y.; Krishnamurthy, S.; Woodward, W.; Alvarez, R.H.; Lucci, A.; Valero, V.; Reuben, J.M.; Meric-Bernstam, F.; et al. Identification of frequent somatic mutations in inflammatory breast cancer. Breast Cancer Res. Treat. 2017, 163, 263–272. [Google Scholar] [CrossRef]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef] [Green Version]
- Marquard, A.M.; Eklund, A.C.; Joshi, T.; Krzystanek, M.; Favero, F.; Wang, Z.C.; Richardson, A.L.; Silver, D.P.; Szallasi, Z.; Birkbak, N.J. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res. 2015, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Srihari, S.; Lal, S.; Gautier, B.; Simpson, P.T.; Khanna, K.K.; Ragan, M.A.; Lê Cao, K.A. Personalised pathway analysis reveals association between DNA repair pathway dysregulation and chromosomal instability in sporadic breast cancer. Mol. Oncol. 2016, 10, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamm, C.A.; Moran, D.; Rao, K.; Trusk, P.B.; Pry, K.; Sausen, M.; Jones, S.; Velculescu, V.E.; Cristofanilli, M.; Bacus, S. Genomic and Immunological Tumor Profiling Identifies Targetable Pathways and Extensive CD8+/PDL1+ Immune Infiltration in Inflammatory Breast Cancer Tumors. Mol. Cancer Ther. 2016, 15, 1746–1756. [Google Scholar] [CrossRef] [Green Version]
- Moslehi, R.; Freedman, E.; Zeinomar, N.; Veneroso, C.; Levine, P.H. Importance of hereditary and selected environmental risk factors in the etiology of inflammatory breast cancer: A case-comparison study. BMC Cancer 2016, 16, 334. [Google Scholar] [CrossRef] [Green Version]
- Rana, H.Q.; Sacca, R.; Drogan, C.; Gutierrez, S.; Schlosnagle, E.; Regan, M.M.; Speare, V.; LaDuca, H.; Dolinsky, J.; Garber, J.E.; et al. Prevalence of germline variants in inflammatory breast cancer. Cancer 2019, 125, 2194–2202. [Google Scholar] [CrossRef]
- Fouad, T.M.; Ueno, N.T.; Yu, R.K.; Ensor, J.E.; Alvarez, R.H.; Krishnamurthy, S.; Lucci, A.; Reuben, J.M.; Yang, W.; Willey, J.S.; et al. Distinct epidemiological profiles associated with inflammatory breast cancer (IBC): A comprehensive analysis of the IBC registry at The University of Texas MD Anderson Cancer Center. PLoS ONE 2018, 13, e0204372. [Google Scholar] [CrossRef]
- Blancato, J.; Singh, B.; Liu, A.; Liao, D.J.; Dickson, R.B. Correlation of amplification and overexpression of the c-myc oncogene in high-grade breast cancer: FISH, in situ hybridisation and immunohistochemical analyses. Br. J. Cancer 2004, 90, 1612–1619. [Google Scholar] [CrossRef]
- Haupt, S.; Buckley, D.; Pang, J.M.; Panimaya, J.; Paul, P.J.; Gamell, C.; Takano, E.A.; Lee, Y.Y.; Hiddingh, S.; Rogers, T.M.; et al. Targeting Mdmx to treat breast cancers with wild-type p53. Cell Death Dis. 2015, 6, e1821. [Google Scholar] [CrossRef] [Green Version]
- Miranda, P.J.; Buckley, D.; Raghu, D.; Pang, J.B.; Takano, E.A.; Vijayakumaran, R.; Teunisse, A.F.; Posner, A.; Procter, T.; Herold, M.J.; et al. MDM4 is a rational target for treating breast cancers with mutant p53. J. Pathol. 2017, 241, 661–670. [Google Scholar] [CrossRef]
- Gao, C.; Xiao, G.; Piersigilli, A.; Gou, J.; Ogunwobi, O.; Bargonetti, J. Context-dependent roles of MDMX (MDM4) and MDM2 in breast cancer proliferation and circulating tumor cells. Breast Cancer Res. 2019, 21, 5. [Google Scholar] [CrossRef]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Rypens, C.; Marsan, M.; Van Berckelaer, C.; Billiet, C.; Melis, K.; Lopez, S.P.; van Dam, P.; Devi, G.R.; Finetti, P.; Ueno, N.T.; et al. Inflammatory breast cancer cells are characterized by abrogated TGFβ1-dependent cell motility and SMAD3 activity. Breast Cancer Res. Treat. 2020, 180, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Vlachou, T.; Luzi, L.; Melloni, G.; Mazzarella, L.; D’Elia, E.; Aobuli, X.; Pasi, C.E.; Reavie, L.; Bonetti, P.; et al. p53 Loss in Breast Cancer Leads to Myc Activation, Increased Cell Plasticity, and Expression of a Mitotic Signature with Prognostic Value. Cell Rep. 2019, 26, 624–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; McClendon, A.K.; Franco, J.; Ertel, A.; Fortina, P.; Witkiewicz, A.K. RB loss contributes to aggressive tumor phenotypes in MYC-driven triple negative breast cancer. Cell Cycle 2015, 14, 109–122. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Jin, F.; Yu, Z.; Zhao, L.; Wang, L.; Bai, X.; Zhao, H.; Yao, W.; Mi, X.; Wang, E.; et al. MYC overexpression and poor prognosis in sporadic breast cancer with BRCA1 deficiency. Tumour Biol. 2013, 34, 3945–3958. [Google Scholar] [CrossRef]
- Gay-Bellile, M.; Véronèse, L.; Combes, P.; Eymard-Pierre, E.; Kwiatkowski, F.; Dauplat, M.M.; Cayre, A.; Privat, M.; Abrial, C.; Bignon, Y.J.; et al. TERT promoter status and gene copy number gains: Effect on TERT expression and association with prognosis in breast cancer. Oncotarget 2017, 8, 77540–77551. [Google Scholar] [CrossRef]
- Knappskog, S.; Berge, E.O.; Chrisanthar, R.; Geisler, S.; Staalesen, V.; Leirvaag, B.; Yndestad, S.; de Faveri, E.; Karlsen, B.O.; Wedge, D.C.; et al. Concomitant inactivation of the p53- and pRB- functional pathways predicts resistance to DNA damaging drugs in breast cancer in vivo. Mol. Oncol. 2015, 9, 1553–1564. [Google Scholar] [CrossRef] [Green Version]
- Van Heetvelde, M.; Van Bockstal, M.; Poppe, B.; Lambein, K.; Rosseel, T.; Atanesyan, L.; Deforce, D.; Van Den Berghe, I.; De Leeneer, K.; Van Dorpe, J.; et al. Accurate detection and quantification of epigenetic and genetic second hits in BRCA1 and BRCA2-associated hereditary breast and ovarian cancer reveals multiple co-acting second hits. Cancer Lett. 2018, 425, 125–133. [Google Scholar] [CrossRef]
- Tuna, M.; Smid, M.; Zhu, D.; Martens, J.W.; Amos, C.I. Association between acquired uniparental disomy and homozygous mutations and HER2/ER/PR status in breast cancer. PLoS ONE 2010, 5, e0015094. [Google Scholar] [CrossRef]
- Martín-Sánchez, E.; Mendaza, S.; Ulazia-Garmendia, A.; Monreal-Santesteban, I.; Blanco-Luquin, I.; Córdoba, A.; Vicente-García, F.; Pérez-Janices, N.; Escors, D.; Megías, D.; et al. CHL1 hypermethylation as a potential biomarker of poor prognosis in breast cancer. Oncotarget 2017, 8, 15789–15801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.H.; Ma, Q.; Shi, Y.H.; Ge, J.; Zhao, H.M.; Li, S.F.; Tong, Z.S. CHL1 is involved in human breast tumorigenesis and progression. Biochem. Biophys. Res. Commun. 2013, 438, 433–438. [Google Scholar] [CrossRef]
- Morrow, R.J.; Etemadi, N.; Yeo, B.; Ernst, M. Challenging a Misnomer? The Role of Inflammatory Pathways in Inflammatory Breast Cancer. Mediat. Inflamm. 2017, 2017, 4754827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, K.; Pandey, N.B.; Popel, A.S. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res. 2018, 20, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patin, E.C.; Soulard, D.; Fleury, S.; Hassane, M.; Dombrowicz, D.; Faveeuw, C.; Trottein, F.; Paget, C. Type I IFN Receptor Signaling Controls IL7-Dependent Accumulation and Activity of Protumoral IL17A-Producing γδT Cells in Breast Cancer. Cancer Res. 2018, 78, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Maciejczyk, A.; Szelachowska, J.; Szynglarewicz, B.; Szulc, R.; Szulc, A.; Wysocka, T.; Jagoda, E.; Lage, H.; Surowiak, P. CD46 Expression is an unfavorable prognostic factor in breast cancer cases. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 540–546. [Google Scholar] [CrossRef]
- Ikeda, J.; Morii, E.; Liu, Y.; Qiu, Y.; Nakamichi, N.; Jokoji, R.; Miyoshi, Y.; Noguchi, S.; Aozasa, K. Prognostic significance of CD55 expression in breast cancer. Clin. Cancer Res. 2008, 14, 4780–4786. [Google Scholar] [CrossRef] [Green Version]
- Mamidi, S.; Cinci, M.; Hasmann, M.; Fehring, V.; Kirschfink, M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol. Oncol. 2013, 7, 580–594. [Google Scholar] [CrossRef]
- Przybytkowski, E.; Lenkiewicz, E.; Barrett, M.T.; Klein, K.; Nabavi, S.; Greenwood, C.M.; Basik, M. Chromosome-breakage genomic instability and chromothripsis in breast cancer. BMC Genom. 2014, 15, 579. [Google Scholar] [CrossRef] [Green Version]
- Parris, T.Z.; Rönnerman, E.W.; Engqvist, H.; Biermann, J.; Truvé, K.; Nemes, S.; Forssell-Aronsson, E.; Solinas, G.; Kovács, A.; Karlsson, P.; et al. Genome-wide multi-omics profiling of the 8p11-p12 amplicon in breast carcinoma. Oncotarget 2018, 9, 24140–24154. [Google Scholar] [CrossRef] [Green Version]
- Luijten, M.N.H.; Lee, J.X.T.; Crasta, K.C. Mutational game changer: Chromothripsis and its emerging relevance to cancer. Mutat. Res. 2018, 777, 29–51. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Kumar, N.; Bagheri, H.C.; von Mering, C.; Robinson, M.D.; Baudis, M. Chromothripsis-like patterns are recurring but heterogeneously distributed features in a survey of 22,347 cancer genome screens. BMC Genom. 2014, 15, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, F.; Guedj, M.; Jones, N.; Sfar, S.; Brouste, V.; Elarouci, N.; Banneau, G.; Orsetti, B.; Primois, C.; de Lara, C.T.; et al. An array CGH based genomic instability index (G2I) is predictive of clinical outcome in breast cancer and reveals a subset of tumors without lymph node involvement but with poor prognosis. BMC Med. Genom. 2012, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [Green Version]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [Green Version]
- Sansregret, L.; Vanhaesebroeck, B.; Swanton, C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, A.E.; Edge, S.B.; Hortobagyi, G.N. Eight edition of the AJCC Cancer Staging Manual: Breast Cancer. Ann. Surg. Oncol. 2018, 25, 1783–1785. [Google Scholar] [CrossRef]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [Green Version]
- Wolff, A.C.; Hammond, M.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W.; et al. Human Epidermal Growth Factor Receptor 2 Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2018, 36, 2105–2122. [Google Scholar] [CrossRef] [Green Version]
- Villacis, R.A.; Basso, T.R.; Canto, L.M.; Pinheiro, M.; Santiago, K.M.; Giacomazzi, J.; de Paula, C.A.A.; Carraro, D.M.; Ashton-Prolla, P.; Achatz, M.I.; et al. Rare germline alterations in cancer-related genes associated with the risk of multiple primary tumor development. J. Mol. Med. 2017, 95, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.J.; do Canto, L.M.; Rogatto, S.R.; Baumbach, J. CoNVaQ: A web tool for copy number variation-based association studies. BMC Genom. 2018, 19, 369. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Liu, J.; Ouyang, L.; Chen, Y.; Liu, B.; Cai, H. CTLPScanner: A web server for chromothripsis-like pattern detection. Nucleic Acids Res. 2016, 44, W252–W258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, M.C.; Marconi, G.; Feenstra, J.D.M.; Fonzi, E.; Papayannidis, C.; Ghelli Luserna di Rorá, A.; Padella, A.; Solli, V.; Franchini, E.; Ottaviani, E.; et al. Chromothripsis in acute myeloid leukemia: Biological features and impact on survival. Leukemia 2018, 32, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- The Genome Aggregation Database (GnomAD). Available online: https://gnomad.broadinstitute.org (accessed on 25 May 2020).
- Naslavsky, M.S.; Yamamoto, G.L.; de Almeida, T.F. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- ClinVar. Available online: http://www.clinvar.com (accessed on 25 May 2020).
- Clinvitae. Available online: http://clinvitae.invitae.com (accessed on 25 May 2020).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- cBioPortal for Cancer Genomics. Available online: https://www.cbioportal.org (accessed on 25 May 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Number of Patients (%) 2 |
|---|---|
| Age (years) | |
| <50 | 10 (29) |
| ≥50 | 24 (71) |
| Family history of cancer | |
| No | 9 (26) |
| Yes | 23 (68) |
| Breast and/or ovarian cancer | 12 (35) |
| Unknown | 2 (6) |
| Body Mass Index (Kg/m2) | |
| ≤24.9 (Normal) | 7 (20) |
| 25 to 30 (Overweight) | 5 (15) |
| >30 (Obese) | 20 (59) |
| Unknown | 2 (6) |
| Histological grade 1 | |
| I | 1 (3) |
| II | 14 (41) |
| III | 19 (56) |
| Clinical stage | |
| III | 21 (62) |
| IV | 13 (38) |
| Hormone receptor (ER or PR) /HER2 status | |
| +/+ | 0 |
| +/− | 18 (53) |
| −/+ | 5 (15) |
| TNBC | 11 (32) |
| Distant metastases | |
| No | 8 (24) |
| Yes | 26 (76) |
| At diagnosis (Stage IV) | 13 (38) |
| During follow-up | 13 (38) |
| Survival status | |
| Alive | 7 (20) |
| Dead | 25 (74) |
| Loss of follow-up | 2 (6) |
| Cases | TP53 | PIK3CA | HR gGenes | MMR Genes |
|---|---|---|---|---|
| IBC1 | c.3140A>G;p.His1047Arg | |||
| IBC2 | c.1633G>A; p.Glu545Lys | |||
| IBC4 | ATM (c.3887C > G; p.Pro1296Arg) PG, Loss | |||
| IBC5 | BRCA2 (c.4963_4964insA; p.Tyr1655Ter) Loss | |||
| IBC6 | ||||
| IBC7 | BRCA2 (C.2806_2809delAAAC; p.Ala938Profs) cnLOH | MLH1 (c.2146G > A; p.Val716Met) PG, Loss | ||
| IBC8 | c.512A > G; p.Glu171Gly PG, Loss | |||
| IBC9 | BRCA2 (c.5682C > G; p.Tyr1894Ter) Loss RAD51B (c.728A > G; p.Lys243Arg) cnLOH POLD1 (c.1055G > A; p.Arg352His) Gain | |||
| IBC10 | BRCA2 (c.2T > G; p.Met1?) | |||
| IBC11 | c.309C > A; p.Tyr103Ter Gain and cnLOH | BRCA2 (c.316+1G >T) Gain and cnLOH RAD51B (c.315+8A > G) PG, Gain and cnLOH | MSH6 (c.3961A > G; p.Arg1321Gly) PG | |
| IBC12 | c.659A > G; p.Tyr220Cys PG, cnLOH | RAD51D (c.899G > A; p.Arg300Gln) PG, Loss | PMS2 (c.2186_2187delTC; p.Leu729Glnfs) PG, Gain | |
| IBC13 | c.712T > G; p.Cys238Gly PG | BRCA2 (c.6988A > G; p.Ile2330Val) PG | ||
| IBC14 | c.586C > T; p.Arg196Ter PG, Loss | BRCA2 (c.8869C > T; p.Gln2957Ter) Loss | ||
| IBC15 | PALB2 (c.43G > T; p.Glu15Ter) cnLOH | MSH3 (c.1567G > A; p.Glu523Lys) PG MSH3 (c.1571A > C; p.Asn524Thr) PG | ||
| IBC16 | c.856G > A; p.Glu286Lys Loss and cnLOH | MSH3 (c.2436-5C > G Gain | ||
| IBC18 | c.626_627delGA; p.Arg209Lysfs PG, Loss | c.3140A > T; p.His1047Leu | BRCA2 (c.9227G > T; p.Gly3076Val) Loss | |
| IBC20 | c.578A > T; p.His193Leu PG c.560-2A > C PG, Loss | c.1035T > A; p.Asn345Lys | BRCA1 (c.3858delT; p.Ser1286Argfs) PG, Loss | MLH3 (c.2638C > G; p.Leu880Val) PG |
| IBC22 | c.731G > A; p.Gly244Asp Loss and cnLOH | c.3140A > G; p.His1047Arg | MUS81 (c.416G > A; p.Arg139Gln) PG | PMS2 (c.2383G > A; p.Asp795Asn) PG |
| IBC24 | c.844C > T; p.Arg282Trp PG, Loss | MSH6 (c.1406A > G; p.Tyr469Cys) PG | ||
| IBC25 | ||||
| IBC26 | c.542G > A; p.Arg181His PG, Loss: |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faldoni, F.L.C.; Villacis, R.A.R.; Canto, L.M.; Fonseca-Alves, C.E.; Cury, S.S.; Larsen, S.J.; Aagaard, M.M.; Souza, C.P.; Scapulatempo-Neto, C.; Osório, C.A.B.T.; et al. Inflammatory Breast Cancer: Clinical Implications of Genomic Alterations and Mutational Profiling. Cancers 2020, 12, 2816. https://doi.org/10.3390/cancers12102816
Faldoni FLC, Villacis RAR, Canto LM, Fonseca-Alves CE, Cury SS, Larsen SJ, Aagaard MM, Souza CP, Scapulatempo-Neto C, Osório CABT, et al. Inflammatory Breast Cancer: Clinical Implications of Genomic Alterations and Mutational Profiling. Cancers. 2020; 12(10):2816. https://doi.org/10.3390/cancers12102816
Chicago/Turabian StyleFaldoni, Flávia L. C., Rolando A. R. Villacis, Luisa M. Canto, Carlos E. Fonseca-Alves, Sarah S. Cury, Simon J. Larsen, Mads M. Aagaard, Cristiano P. Souza, Cristovam Scapulatempo-Neto, Cynthia A. B. T. Osório, and et al. 2020. "Inflammatory Breast Cancer: Clinical Implications of Genomic Alterations and Mutational Profiling" Cancers 12, no. 10: 2816. https://doi.org/10.3390/cancers12102816