Management of Small Bowel Neuroendocrine Tumors


Abstract
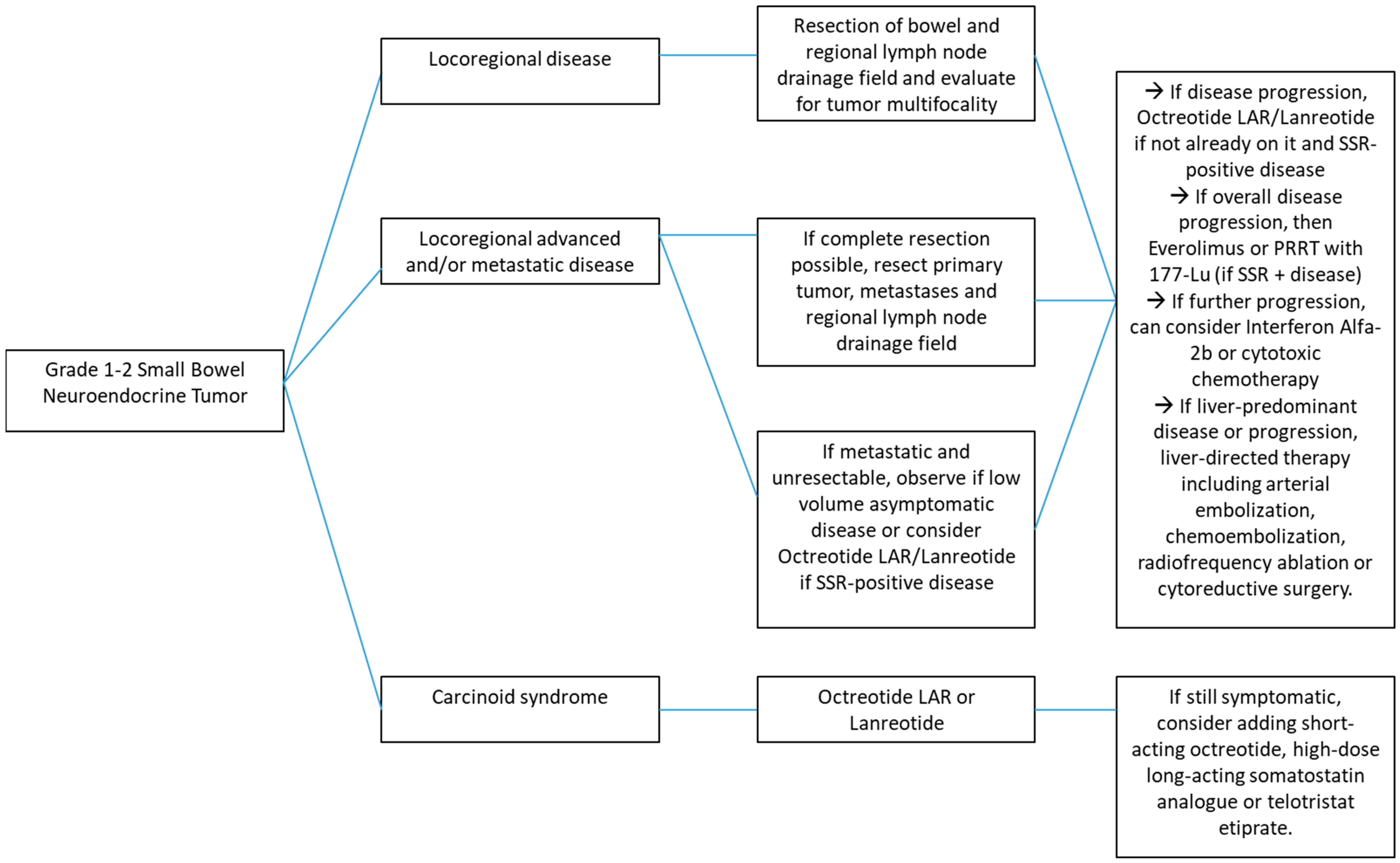
:1. Introduction
2. Locoregional Disease
3. Synchronous Primary and Metastatic Disease
4. Hepatic Metastases
5. Other Surgical Considerations
6. Systemic Therapy for Metastatic or Unresectable Disease
7. Carcinoid Syndrome and Carcinoid Heart Disease
8. Management of Small Bowel Neuroendocrine Carcinoma
9. Conclusions
Author Contributions
Conflicts of Interest
References
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.N.; Rashid, A.; et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. 2008, 26, 3063. [Google Scholar] [CrossRef] [PubMed]
- Ohike, N.; Adsay, N.V.; La Rosa, S. Mixed neuroendocrine-non-neuroendocrine neoplasms. In WHO Classification of Tumours of Endocrine Organs, 4th ed.; Lloyd, R.V., Osamura, R.Y., Kloppel, G., Rosai, J., Eds.; IARC Press: Lyon, France, 2017; p. 238. [Google Scholar]
- Woltering, E.A.; Bergsland, E.K.; Beyer, D.T.; O’Dorisio, T.M.; Rindi, G.; Klimstra, D.S.; Tang, L.H.; Reidy-Lagunes, D.; Strosberg, J.R.; Wolin, E.M.; et al. Neuroendocrine tumors of the jejunum and ileum. In AJCC Cancer Staging Manual, 8th ed.; Amin, M.B., Ed.; AJCC: Chicago, IL, USA, 2017; p. 375. [Google Scholar]
- Barsouk, A.; Rawla, P.; Barsouk, A.; Thandra, K.C. Epidemiology of Cancers of the Small Intestine: Trends, Risk Factors and Prevention. Med. Sci. (Basel) 2019, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Auernhammer, C.J.; Spitzweg, C.; Angele, M.K.; Boeck, S.; Grossman, A.; Nölting, S.; Ilhan, H.; Knösel, T.; Mayerle, J.; Reincke, M.; et al. Advanced neuroendocrine tumours of the small intestine and pancreas: Clinical developments, controversies, and future strategies. Lancet Diabetes Endocrinol. 2018, 6, 404–415. [Google Scholar] [CrossRef]
- Moris, D.; Ntanasis-Stathopoulos, I.; Tsilimigras, D.I.; Vagios, S.; Karamitros, A.; Karaolanis, G.; Griniatsos, J.; Papalampros, A.; Papaconstantinou, I.; Glantzounis, G.K.; et al. Update on Surgical Management of Small Bowel Neuroendocrine Tumors. Anticancer Res. 2018, 38, 1267–1278. [Google Scholar] [PubMed] [Green Version]
- Scott, A.T.; Howe, J.R. Management of small bowel neuroendocrine tumors. J. Oncol. Pract. 2018, 14, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Rinke, A.; Müller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Bläker, M.; et al. Placeboo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.E.; Pavel, M.; Jarosław, B.Ć.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. NEJM 2014, 371, 224–233. [Google Scholar] [CrossRef]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef]
- Strosberg, J.; Wolin, G.E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; Bushnell, D.; et al. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. NEJM 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Singh, S.; Asa, S.L.; Dey, C.; Kennecke, H.; Laidley, D.; Law, C.; Asmis, T.; Chan, D.; Ezzat, S.; Goodwin, R.; et al. Diagnosis and management of gastrointestinal neuroendocrine tumors: An evidence-based Canadian consensus. Cancer Treat. Rev. 2016, 47, 32–45. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Halfdanarson, T.R.; Bellizzi, A.M.; Chan, J.A.; Dillon, J.S.; Heaney, A.P.; Kunz, P.L.; O’Dorisio, T.M.; Salem, R.; Segelov, E.; et al. The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Medical Management of Midgut Neuroendocrine Tumors. Pancreas 2017, 46, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Howe, J.R.; Cardona, K.; Fraker, D.L.; Kebebew, E.; Untch, B.R.; Wang, Y.Z.; Law, C.H.; Liu, E.H.; Kim, M.K.; Menda, Y.; et al. The Surgical Management of Small Bowel Neuroendocrine Tumors: Consensus Guidelines of the North American Neuroendocrine Tumor Society. Pancreas 2017, 46, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Kulke, M.H.; Goldner, W.S.; Benson, A.B.; Bergsland, E.; Berlin, J.D.; Blaszowsky, L.S.; Chan, J.; Eads, J.; Engstrom, P.F.; et al. NCCN Clinical Practice Guidelines in Oncology: Neuroendocrine and Adrenal Tumors. J. Natl. Compr. Canc. Netw. 2018, 16, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Niederle, B.; Pape, U.F.; Costa, F.; Gross, D.; Kelestimur, F.; Knigge, U.; Öberg, K.; Pavel, M.; Perren, A.; Toumpanakis, C.; et al. ENETS Consensus Guidelines Update for Neuroendocrine Neoplasms of the Jejunum and Ileum. Neuroendocrinology 2016, 103, 125–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavel, M.; O’Toole, D.; Costa, F.; Capdevila, J.; Gross, D.; Kianmanesh, R.; Krenning, E.; Knigge, U.; Salazar, R.; Pape, U.F.; et al. ENETS Consensus Guidelines Update for the Management of Distant Metastatic Disease of the Intestinal, Pancreatic, Bronchial Neuroendocrine Neoplasms and NEN of Unknown Primary Site. Neuroendocrinology 2016, 103, 172–185. [Google Scholar] [CrossRef]
- Rorstad, O. Prognoostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J. Surg. Oncol. 2005, 89, 151. [Google Scholar] [CrossRef] [PubMed]
- Cives, M.; Anaya, D.A.; Soares, H.; Coppola, D.; Strosberg, J. Analysis of Postoperative Recurrence in Stage I-III Midgut Neuroendocrine Tumors. J. Natl. Cancer Inst. 2018, 110, 282–289. [Google Scholar] [CrossRef]
- Capurso, G.; Rinzivillo, M.; Bettini, R.; Boninsegna, L.; Delle Fave, G.; Falconi, M. Systematic review of resection of primary midgut carcinoid tumour in patients with unresctable liver metastases. Br. J. Surg. 2012, 99, 1480–1486. [Google Scholar] [CrossRef]
- Almond, L.M.; Hodson, J.; Ford, S.J.; Gourevitch, D.; Roberts, K.J.; Shah, T.; Isaac, J.; Desai, A. Role of Palliative Resection of the Primary Tumour in Advanced Pancreatic and Small Intestinal Neuroendocrine Tumours: A Systematic Review and Meta-analysis. Eur. J. Surg. Oncol. 2017, 43, 1808–1815. [Google Scholar] [CrossRef]
- Daskalakis, K.; Karakatsanis, A.; Hessman, O.; Stuart, H.C.; Welin, S.; Tiensuu Janson, E.; Öberg, K.; Hellman, P.; Norlén, O.; Stålberg, P. Association of a Prophylactic Surgical Approach to Stage IV Small Intestinal Neuroendocrine Tumors with Survival. JAMA Oncol. 2018, 4, 183. [Google Scholar] [CrossRef]
- Chamberlain, R.S.; Canes, D.; Brown, K.T.; Saltz, L.; Jarnagin, W.; Fong, Y.; Blumgart, L.H. Hepatic neuroendocrine metastases: Does intervention alter outcomes? J. Am. Coll. Surg. 2000, 190, 432. [Google Scholar] [CrossRef]
- Elias, D.; Lasser, P.; Ducreux, M.; Duvillard, P.; Ouellet, J.F.; Dromain, C.; Schlumberger, M.; Pocard, M.; Boige, V.; Miquel, C.; et al. Liver resection (and associated extrahepatic resections) for metastatic well-differentiated endocrine tumors: A 15-year single center prospective study. Surgery 2003, 133, 375. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, J.M.; Heywood, G.; Rubin, J.; Ilstrup, D.M.; Nagorney, D.M.; Que, F.G. Surgical treatment of neuroendocrine metastases to the liver: A plea for resection to increase survival. J. Am. Coll. Surg. 2003, 197, 29. [Google Scholar] [CrossRef]
- Osborne, D.A.; Zervos, E.E.; Strosberg, J.; Boe, B.A.; Malafa, M.; Rosemurgy, A.S.; Yeatman, T.J.; Carey, L.; Duhaine, L.; Kvols, L.K. Improved outcome with cytoreduction versus embolization for symptomatic hepatic metastases of carcinoid and neuroendocrine tumors. Ann. Surg. Oncol. 2006, 13, 572. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Turner, G.; King, B.; Jones, L.; Culliford, D.; McCance, D.; Ardill, J.; Johnston, B.T.; Poston, G.; Rees, M.; et al. Midgut neuroendocrine tumours with liver metastases: Results of the UKINETS study. Endocr. Relat. Cancer 2009, 16, 885. [Google Scholar] [CrossRef] [PubMed]
- Glazer, E.S.; Tseng, J.F.; Al-Refaie, W.; Solorzano, C.C.; Liu, P.; Willborn, K.A.; Abdalla, E.K.; Vauthey, J.N.; Curley, S.A. Long-term survival after surgical management of neuroendocrine hepatic metastases. HPB (Oxf.) 2010, 12, 427. [Google Scholar] [CrossRef] [PubMed]
- Mayo, S.C.; de Jong, M.C.; Pulitano, C.; Clary, B.M.; Reddy, S.K.; Gamblin, T.C.; Celinksi, S.A.; Kooby, D.A.; Staley, C.A.; Stokes, J.B.; et al. Surgical management of hepatic neuroendocrine tumor metastasis: Results from an international multi-institutional analysis. Ann. Surg. Oncol. 2010, 17, 3129. [Google Scholar] [CrossRef]
- Saxena, A.; Chua, T.C.; Sarkar, A.; Chu, F.; Liauw, W.; Zhao, J.; Morris, D.L. Progression and survival results after radical hepatic metastasectomy of indolent advanced neuroendocrine neoplasms (NENs) supports an aggressive surgical approach. Surgery 2011, 149, 209. [Google Scholar] [CrossRef]
- Gaujoux, S.; Gonen, M.; Tang, L.; Klimstra, D.; Brennan, M.F.; D’Angelica, M.; Dematteo, R.; Allen, P.J.; Jarnagin, W.; Fong, Y. Synchronous resection of primary and liver metastases for neuroendocrine tumors. Ann. Surg. Oncol. 2012, 19, 4270. [Google Scholar] [CrossRef]
- Bagante, F.; Spolverato, G.; Merath, K.; Postlewait, L.M.; Poultsides, G.A.; Mullen, M.G.; Bauer, T.W.; Fields, R.C.; Lamelas, J.; Marques, H.P.; et al. Neuroendocrine liver metastases: The chance to be cured after liver surgery. J. Surg. Oncol. 2017, 115, 679. [Google Scholar] [CrossRef]
- Mayo, S.C.; de Jong, M.C.; Bloomston, M.; Pulitano, C.; Clary, B.M.; Reddy, S.K.; Clark Gamblin, T.; Celinski, S.A.; Kooby, D.A.; Staley, C.A.; et al. Surgery versus intra-arterial therapy for neuroendocrine liver metastasis: A multicenter international analysis. Ann. Surg. Oncol. 2011, 18, 3657–3665. [Google Scholar] [CrossRef] [PubMed]
- Norlén, O.; Stålberg, P.; Zedenius, J.; Hellman, P. Outcome after resection and radiofrequency ablation of liver metastases from small intestinal neuroendocrine tumours. Br. J. Surg. 2013, 100, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Akyildiz, H.Y.; Mitchell, J.; Milas, M.; Siperstein, A.; Berber, E. Laparoscopic radiofrequency thermal ablation of neuroendocrine hepatic metastases: Long-term follow-up. Surgery 2010, 148, 1288–1293. [Google Scholar] [CrossRef]
- Drougas, J.G.; Anthony, L.B.; Blair, T.K.; Lopez, R.R.; Wright, J.K., Jr.; Chapman, W.C.; Webb, L.; Mazer, M.; Meranze, S.; Pinson, C.W. Hepatic Artery Chemoembolization for Management of Patients with Advanced Metastatic Carcinoid Tumors. Am. J. Surg. 1998, 175, 408. [Google Scholar] [CrossRef]
- Gupta, S.; Yao, J.C.; Ahrar, K.; Wallace, M.J.; Morello, F.A.; Madoff, D.C.; Murthy, R.; Hicks, M.E.; Ajani, J.A. Hepatic Artery Embolization and Chemoembolization for Treatment of Patients with Metastatic Carcinoid Tumors: The M.D. Anderson Experience. Cancer J. 2003, 9, 261. [Google Scholar] [CrossRef]
- Kitano, M.; Davidson, G.W.; Shirley, L.A.; Schmidt, C.R.; Guy, G.E.; Khabiri, H.; Dowell, J.D.; Shah, M.H.; Bloomston, M. Transarterial Chemoembolization for Metastatic Neuroendorine Tumors with Massive Hepatic Tumor Burden: Is the Benefit Worth the Risk? Ann. Surg. Oncol. 2016, 23, 4008–4015. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.; Bester, L.; Salem, R.; Sharma, R.A.; Parks, R.W.; Ruszniewski, P.; NET-Liver-Metastases Consensus Conference. Role of intra-arterial therapies in metastatic neuroendocrine tumours (NET): Guidelines from the NET-Liver-Metastases Consensus Conference. HPB (Oxf.) 2015, 17, 29. [Google Scholar] [CrossRef]
- Norlén, O.; Hessman, O.; Stålberg, P.; Akerström, G.; Hellman, P. Prophylactic Cholecystectomy in Midgut Carcinoid Patients. World J. Surg. 2010, 34, 1361–1367. [Google Scholar] [CrossRef]
- Trendle, M.C.; Moertel, C.G.; Kvols, L.K. Incidence and Morbidity of Cholelithiasis in Patients REceiving Chronic Octreotide for Metastatic Carcinoid and Malignant Islet Cell Tumors. Cancer 1997, 79, 830–834. [Google Scholar] [CrossRef]
- Brighi, N.; Lamberti, G.; Maggio, I.; Manuzzi, L.; Ricci, C.; Casadei, R.; Santini, D.; Mosconi, C.; Lisotti, A.; Ambrosini, V.; et al. Biliary Stone Disease in Patients Receiving Somatostatin Analogs for Neuroendocrine Neoplasms: A Retrospective Observational Study. Dig Liver Dis. 2019, 51, 689–694. [Google Scholar] [CrossRef]
- Le Treut, Y.P.; Grégoire, E.; Klempnauer, J.; Belghiti, J.; Jouve, E.; Lerut, J.; Castaing, D.; Soubrane, O.; Boillot, O.; Mantion, G.; et al. Liver transplantation for neuroendocrine tumors in Europe—Results and trends in patient selection: A 213-case European liver transplant registry study. Ann. Surg. 2013, 257, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Gedaly, R.; Daily, M.F.; Davenport, D.; McHugh, P.P.; Koch, A.; Angulo, P.; Hundley, J.C. Liver transplantation for the treatment of liver metastases from neuroendocrine tumors: An analysis of the UNOS database. Arch. Surg. 2011, 146, 953–958. [Google Scholar] [CrossRef] [PubMed]
- Puccini, A.; Battaglin, F.; Lenz, H.J. Management of Advanced Small Bowel Cancer. Curr. Treat. Options Oncol. 2018, 19, 69. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Hörsch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M.; et al. Everolimus Plus Octreotide Long-Acting Repeatble for the Treatment of Advanced Neuroendocrine Tumours Associated with Carcinoid Syndrome (RADIANT-2): A Randomised, Placebo-Controlled, Phase 3 study. Lancet 2011, 378, 2005–2012. [Google Scholar] [CrossRef]
- Strosberg, J.; Wolin, E.; Chasen, B.; Kulke, M.; Bushnell, D.; Caplin, M.; Baum, R.P.; Hobday, T.; Hendifar, A.; Lopera Sierra, M.; et al. First Update on Overall Survival, Progresion-Free Survival, and Health-Related Time-to-Deterioration Quality of Life from the NETTER-1 Study: 177-Lu-Dotatate vs High Dose Octreotide in Progressive Midgut Neuroendocrine Tumors [Abstract 4099]. In Proceedings of the 2018 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, USA, 1–5 June 2018. [Google Scholar]
- Rinke, A.; Neary, M.P.; Eriksson, J.; Hunger, M.; Doan, T.; Karli, D.; Arnold, R. Health-related Quality of Life for Octreotide Long-Acting vs. Placebo in Patients with Metastatic Midgut Neuroendocrine Tumors in the Phase 3 PROMID Trial. Neuroendocrinology 2019, 109, 141–151. [Google Scholar] [CrossRef]
- Pavel, M.E.; Singh, S.; Strosberg, J.R.; Bubuteishvili-Pacaud, L.; Degtyarev, E.; Neary, M.P.; Carnaghi, C.; Tomasek, J.; Wolin, E.; Raderer, M.; et al. Health-related Quality of Life for Everolimus versus Placebo in Patients with Advanced, Non-Functional, Well-Differentiated Gastrointestinal of Lung Neuroendocrine Tumours (RADIANT-4): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1411–1422. [Google Scholar]
- Strosberg, J.; Wolin, E.; Chasen, B.; Kulke, M.; Bushnell, D.; Caplin, M.; Baum, R.P.; Kunz, P.; Hobday, T.; Hendifar, A.; et al. Health-related Quality of Life in Patients with Progressive Midgust Neuroendocrine Tumors Treated with 177-Lu-Dotatate in the Phase III NETTER-1 Trial. J. Clin. Oncol. 2018, 36, 2578–2584. [Google Scholar] [CrossRef]
- Di Bartolomeo, M.; Bajetta, E.; Buzzoni, R.; Mariani, L.; Carnaghi, C.; Somma, L.; Zilembo, N.; di Leo, A. Clinicall efficacy of octreotide in the treatment of metastatic neuroendocrine tumors. A study by the Italian Trials in Medical Oncology Group. Cancer 1996, 77, 402. [Google Scholar] [CrossRef]
- Faiss, S.; Pape, U.F.; Böhmig, M.; Dörffel, Y.; Mansmann, U.; Golder, W.; Riecken, E.O.; Wiedenmann, B.; International Lanreotide and Interferon Alfa Study Group. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors—The Internaional Lanreotide and Interferon Alfa Study Group. J. Clin. Oncol. 2003, 21, 2689. [Google Scholar]
- Schöber, C.; Schmoll, E.; Schmoll, H.J.; Poliwoda, H.; Schuppert, F.; Stahl, M.; Bokemeyer, C.; Wilke, H.; Weiss, J. Antitumour effect and symptomatic control with interferon aplha 2b in patients with endocrine active tumours. Eur. J. Cancer 1992, 28, 1664. [Google Scholar] [CrossRef]
- Biesma, B.; Willemse, P.H.; Mulder, N.H.; Verschueren, R.C.; Kema, I.P.; de Bruijn, H.W.; Postmus, P.E.; Sleijfer, D.T.; de Vries, E.G. Recombinant interferon aplha 2b in patients with metastatic apudomas: Effect on tumours and tumour markers. Br. J. Cancer 1992, 66, 850. [Google Scholar] [CrossRef] [PubMed]
- Bajetta, E.; Zilembo, N.; Di Bartolomeo, M.; Di Leo, A.; Pilotti, S.; Bochicchio, A.M.; Castellani, R.; Buzzoni, R.; Celio, L.; Dogliotti, L.; et al. Treatment of metastatic carcinoid and other neuroendocrine tumors with recombinant interferon aplha 2a: A Study by the Italian Trials in Medical Oncology Group. Cancer 1993, 72, 3099. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Vermeulen, P.B.; Fierens, H.; De Schepper, B.; Corthouts, B.; Van Oosterom, A.T. Long-term results of continuous treatment with recombinant interferon-alpha in patients with metastatic carcinoid tumors—An antiangiogenic effect? Anticancer Drugs 1996, 7, 175. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Rubin, J.; Kvols, L.K. Therapy of metastatic carcinoid tumor and the malignant carcinoid syndrome with recombinant leukocyte A interferon. J. Clin. Oncol. 1989, 7, 865. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, L.E.; Schrumpf, E.; Kolbenstvedt, A.N.; Tausjø, J.; Dolva, L.O. Treatment of malignant metastatic midgut carcinoid tumours with recombinant human alpha2b interferon with or without prior hepatic artery embolization. Scand. J. Gastroenterol. 1989, 24, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Creutzfeldt, W.; Bartsch, H.H.; Jacubaschke, U.; Stöckmann, F. Treatment of gastrointestinal endocrine tumours with interferon-alpha and octreotide. Acta Oncol. 1991, 30, 529. [Google Scholar] [CrossRef] [PubMed]
- Oberg, K.; Eriksson, L. The role of interferons in the management of carcinoid tumours. Br. J. Heamatol. 1991, 79, 74. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Kumpulainen, E.; Gröhn, P. Treatment of metastatic carcinoid tumour with recombinant interferon alfa. Eur. J. Cancer 1992, 28, 1650–1653. [Google Scholar] [CrossRef]
- Janson, E.T.; Rönnblom, L.; Ahlström, H.; Grandér, D.; Alm, G.; Einhorn, S.; Oberg, K. Treatment with alpha-interferon vs alpha-interferon in combination with streptozocin and doxorubicin in patients with malignant carcinoid tumours: A randomized trial. Ann. Oncol. 1992, 3, 635. [Google Scholar] [CrossRef]
- Doberauer, C.; Niederle, N.; Kloke, O.; Kurschel, E.; Schmidt, C.G. Treatment of metastasized carcinoid tumor of the ileum and cecum with recombinant alpha-2b-interferon. Onkologie 1987, 10, 340–344. [Google Scholar]
- Kölby, L.; Persson, G.; Franzén, S.; Ahrén, B. Randomized clinical trial of the effect of interferon alpha on survival in patients with disseminated midgut carcinoid tumours. Br. J. Surg. 2003, 90, 687. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.; Rinke, A.; Klose, K.J.; Müller, H.H.; Wied, M.; Zamzow, K.; Schmidt, C.; Schade-Brittinger, C.; Barth, P.; Moll, R.; et al. Octreotide versus octreotide plus interferon-alpha in endocrine gastroenteropancreatic tumors: A randomized trial. Clin. Gastroenterol. Hepatol. 2005, 3, 761–771. [Google Scholar] [CrossRef]
- Kulke, M.H.; Lenz, H.J.; Meropol, N.J.; Posey, J.; Ryan, D.P.; Picus, J.; Bergsland, E.; Stuart, K.; Tye, L.; Huang, X.; et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J. Clin. Oncol. 2008, 26, 3403. [Google Scholar] [CrossRef] [PubMed]
- Hobday, T.J.; Rubin, J.; Holen, K.; Picus, J.; Donehower, R.; Maples, R.M.; Lloyd, R.; Mahoney, M.; Erlichman, C. MC044h, a Phase II Trial of Sorafenib in Patients with Metastatic Neuroendocrine Tumors (NET): A Phase II Consortium study. J. Clinl. Oncol. 2007, 25 (Suppl. 18), 4504. [Google Scholar]
- Phan, A.T.; Halperin, D.M.; Chan, J.A.; Fogelman, D.R.; Hess, K.R.; Malinowski, P.; Regan, E.; Ng, C.S.; Yao, J.C.; Kulke, M.H. Pazopanib and depot octreotide in advanced, well-differentiated neuroendocrine tumours: A multicentre, single-group, phase 2 study. Lancet Oncol. 2015, 16, 695. [Google Scholar] [CrossRef]
- Grande, E.; Capdevila, J.; Castellano, D.; Teulé, A.; Durán, I.; Fuster, J.; Sevilla, I.; Escudero, P.; Sastre, J.; García-Donas, J.; et al. Pazopanib in pretreated advanced neuroendocrine tumors: A phase II, open-label trial of the Spanish Task Force Group for Neuroendocrine Tumors (GETNE). Ann. Oncol. 2015, 26, 1987. [Google Scholar] [CrossRef]
- Yao, J.C.; Guthrie, K.A.; Moran, C.; Strosberg, J.R.; Kulke, M.H.; Chan, J.A.; LoConte, N.; McWilliams, R.R.; Wolin, E.M.; Mattar, B.; et al. Phase III Prospective Randomized Comparison Trial of Depot Octreotide Plus INterferon Alfa-2b Versus Depot Octreotide Plus Bevacizumab in Patients with Advanced Carcinoid Tumors. J. Clin. Oncol. 2017, 35, 1695–1703. [Google Scholar] [CrossRef]
- Bukowski, R.M.; Tangen, C.M.; Peterson, R.F.; Taylor, S.A.; Rinehart, J.J.; Eyre, H.J.; Rivkin, S.E.; Fleming, T.R.; Macdonald, J.S. Phase II trial of dimethyltriazenoimidazole carboxamide in patients with metastatic carcinoid. A Southwest Oncology Group study. Cancer 1994, 73, 1505. [Google Scholar] [CrossRef]
- Sun, W.; Lipsitz, S.; Catalano, P.; Mailliard, J.A.; Haller, D.G.; Eastern Cooperative Oncology Group. Phase II/III study of doxorubicin with fluorouracil compared with streptozocin with fluorouracil or dacarbazine in the treatment of advanced carcinoid tumors: Eastern Cooperative Oncology Group Study E1281. J. Clin. Oncol. 2005, 23, 4897. [Google Scholar] [CrossRef]
- Ansell, S.M.; Pitot, H.C.; Burch, P.A.; Kvols, L.K.; Mahoney, M.R.; Rubin, J. A Phase II study of high-dose paclitaxel in patients with advanced neuroendocrine tumors. Cancer 2001, 91, 1543. [Google Scholar] [CrossRef]
- Kulke, M.H.; Kim, H.; Stuart, K.; Clark, J.W.; Ryan, D.P.; Vincitore, M.; Mayer, R.J.; Fuchs, C.S. A phase II study of docetaxel in patients with metastatic carcinoid tumors. Cancer Investig. 2004, 22, 353. [Google Scholar] [CrossRef]
- Kulke, M.H.; Kim, H.; Clark, J.W.; Enzinger, P.C.; Lynch, T.J.; Morgan, J.A.; Vincitore, M.; Michelini, A.; Fuchs, C.S. A Phase II trial of gemcitabine for metastatic neuroendocrine tumors. Cancer 2004, 101, 934. [Google Scholar] [CrossRef] [PubMed]
- Medley, L.; Morel, A.N.; Farrugia, D.; Reed, N.; Hayward, N.; Davies, J.M.; Kirichek, O.; Thakker, R.V.; Talbot, D.C. Phase II study of single agent capecitabine in the treatment of metastatic non-pancreatic neuroendocrine tumours. Br. J. Cancer 2011, 104, 1067. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Hanley, J.A. Combination chemotherapy trials in metastatic carcinoid tumor and the malignant carcinoid syndrome. Cancer Clin. Trials 1979, 2, 327. [Google Scholar] [PubMed]
- Engstrom, P.F.; Lavin, P.T.; Moertel, C.G.; Folsch, E.; Douglass, H.O., Jr. Streptozocin plus fluorouracil versus doxorubicin therapy for metastatic carcinoid tumor. J. Clin. Oncol. 1984, 2, 1255. [Google Scholar] [CrossRef] [PubMed]
- Fine, R.L.; Gulati, A.P.; Krantz, B.A.; Moss, R.A.; Schreibman, S.; Tsushima, D.A.; Mowatt, K.B.; Dinnen, R.D.; Mao, Y.; Stevens, P.D.; et al. Capecitabine and temozolomide (CAPTEM) for metastatic, well-differentiated neuroendocrine cancers: The Pancreas Center at Columbia University experience. Cancer Chemother. Pharmacol. 2013, 71, 663. [Google Scholar] [CrossRef] [PubMed]
- Fine, R.L.; Gulati, A.P.; Tsushima, D.; Mowatt, K.B.; Oprescu, A.; Chabot, J.N.B.A. Prospective Phase II Study of Capecitabine and Temozolomide (CAPTEM) for Progressive, Moderately, and Well-Differentiated Metastatic Neuroendocrine Tumor (abstract 179). J. Clin. Oncol. 2014, 32 (Suppl. 3), 179. [Google Scholar] [CrossRef]
- Yordanova, A.; Ahrens, H.; Feldmann, G.; Brossart, P.; Gaertner, F.C.; Fottner, C.; Weber, M.M.; Ahmadzadehfar, H.; Schreckenberger, M.; Miederer, M.; et al. Peptide Receptor Radionuclide Therapy Combined with Chemotherapy in Patients with Neuroendocrine Tumors. Clin. Nucl. Med. 2019, 44, 329–335. [Google Scholar] [CrossRef]
- Kvols, L.K.; Moertel, C.G.; O’Connell, M.J.; Schutt, A.J.; Rubin, J.; Hahn, R.G. Treatment of the malignant carcinoid syndrome: Evaluation of a long-acting somatostatin analogue. NEJM 1986, 315, 663. [Google Scholar] [CrossRef]
- Rubin, J.; Ajani, J.; Schirmer, W.; Venook, A.P.; Bukowski, R.; Pommier, R.; Saltz, L.; Dandona, P.; Anthony, L. Octreotide acetate long-acting formulation versus open-label subcutaneous octreotide acetate in malignant carcinoid syndrome. J. Clin. Oncol. 1999, 17, 600. [Google Scholar] [CrossRef]
- Strosberg, J.; Weber, J.; Feldman, M.; Goldman, J.; Almhanna, K.; Kvols, L. Above-Label doses of Octreotide LAR in patients with metastatic small intestinal carcinoid tumors. Gastrointest. Cancer Res. 2013, 6, 81–85. [Google Scholar] [PubMed]
- Ruszniewski, P.; Ducreux, M.; Chayvialle, J.A.; Blumberg, J.; Cloarec, D.; Michel, H.; Raymond, J.M.; Dupas, J.L.; Gouerou, H.; Jian, R.; et al. Treatment of the carcinoid syndrome with the long-acting somatostatin analogue lanreotide: A prospective study in 39 patients. Gut 1996, 39, 279. [Google Scholar] [CrossRef] [PubMed]
- Ruszniewski, P.; Malka, D. Hepatic artery embolization in the management of advanced digestive endocrine tumors. Digestion 2000, 62, 79. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Choi, J.; Cantor, A.B.; Kvols, L.K. Selective hepatic artery embolization for treatment of patients with metastatic carcinoid and pancreatic endocrine tumors. Cancer Control. 2006, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, B.K.; Larsson, E.G.; Skogseid, B.M.; Löfberg, A.M.; Lörelius, L.E.; Oberg, K.E. Liver embolizations of patients with malignant neuroendocrine gastrointestinal tumors. Cancer 1998, 83, 2293. [Google Scholar] [CrossRef]
- Kulke, M.H.; Hörsch, D.; Caplin, M.E.; Anthony, L.B.; Bergsland, E.; Öberg, K.; Welin, S.; Warner, R.R.; Lombard-Bohas, C.; Kunz, P.L.; et al. Telotristat Ethyl, a Tryptophan Hyroxylase Inhibitor for the Treatment of Carcinoid Syndrome. J. Clin. Oncol. 2017, 35, 14. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.; Klose, K.J.; Wied, M.; Ishaque, N.; Schade-Brittinger, C.; Arnold, R. Combination therapy with octreotide and alpha-interferon: Effect on tumor growth in metastatic endocrine gastroenteropancreatic tumors. Am. J. Gastroenterol. 1999, 94, 1381. [Google Scholar] [PubMed]
- Wolin, E.M.; Jarzab, B.; Eriksson, B.; Walter, T.; Toumpanakis, C.; Morse, M.A.; Tomassetti, P.; Weber, M.M.; Fogelman, D.R.; Ramage, J.; et al. Phase III study of pasireotide long-acting release in patients with metastatic neuroendocrine tumors and carcinoid symptoms refractory to available somatostatin analogue. Drug Des. Devel. Ther. 2015, 9, 5075–5086. [Google Scholar] [CrossRef]
- Rindi, G.; Klimstra, D.S.; Abedi-Ardekani, B.; Asa, S.L.; Bosman, F.T.; Brambilla, E.; Busam, K.J.; de Krijger, R.R.; Dietel, M.; El-Naggar, A.K.; et al. A Common Classification Framework for Neuroendocrine Neoplasms: An International Agency for REsearch on Cancer (IARC) and World Health Organization (WHO) Expert Consensus Proposal. Mod. Parhol. 2018, 31, 1770–1786. [Google Scholar] [CrossRef]
- Dasari, A.; Mehta, K.; Byers, L.; Sorbye, H.; Yao, J.C. Comparative study of lung and extrapulmonary poorly differentiated neuroendocrine carcinoma: A SEER database analysis of 162,983 cases. Cancer 2018, 124, 807. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Coppola, D.; Klimstra, D.S.; Phan, A.T.; Kulke, M.H.; Wiseman, G.A.; Kvols, L.K.; North American Neuroendocrine Tumor Society (NANETS). The NANETS consensus guidelines for the diagnosis and management of poorly differentiated (high-grade) extrapulmonary neuroendocrine carcinomas. Pancreas 2010, 39, 799. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, H.; Strosberg, J.; Baudin, E.; Klimstra, D.S.; Yao, J.C. Gastroenteropancreatic high-grade neuroendocrine carcinoma. Cancer 2014, 120, 2814. [Google Scholar] [CrossRef] [PubMed]
- Haugvik, S.P.; Janson, E.T.; Österlund, P.; Langer, S.W.; Falk, R.S.; Labori, K.J.; Vestermark, L.W.; Grønbæk, H.; Gladhaug, I.P.; Sorbye, H. Surgical Treatment as a Principle for Patients with High-Grade Pancreatic Neuroendocrine Carcinoma: A Nordic Multicenter Comparative Study. Ann. Surg. Oncol. 2016, 23, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Galleberg, R.B.; Knigge, U.; Tiensuu Janson, E.; Vestermark, L.W.; Haugvik, S.P.; Ladekarl, M.; Langer, S.W.; Grønbæk, H.; Österlund, P.; Hjortland, G.O.; et al. Results after surgical treatment of liver metastases in patients with high-grade gastroenteropancreatic neuroendocrine carcinomas. Eur. J. Surg. Oncol. 2017, 43, 1682. [Google Scholar] [CrossRef] [PubMed]
- Vélayoudom-Céphise, F.L.; Duvillard, P.; Foucan, L.; Hadoux, J.; Chougnet, C.N.; Leboulleux, S.; Malka, D.; Guigay, J.; Goere, D.; Debaere, T.; et al. Are G3 ENETS neuroendocrine neoplasms heterogeneous? Endocr. Relat. Cancer 2013, 20, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heetfeld, M.; Chougnet, C.N.; Olsen, I.H.; Rinke, A.; Borbath, I.; Crespo, G.; Barriuso, J.; Pavel, M.; O’Toole, D.; Walter, T.; et al. Characteristics and treatment of patients with G3 gastroenteropancreatic neuroendocrine neoplasms. Endocr. Relat. Cancer 2015, 22, 657–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorbye, H.; Welin, S.; Langer, S.W.; Vestermark, L.W.; Holt, N.; Osterlund, P.; Dueland, S.; Hofsli, E.; Guren, M.G.; Ohrling, K.; et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinomas (WHO G3), the NORDIC NEC study. Ann. Oncol. 2013, 24, 152–160. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). Identifier NCT02595424, Cisplatin, Carboplatin and Etoposide or Temozolomide and Capecitabine in Treating Patients with Neuroendocrine Carcinoma of the Gastrointestinal Tract or Pancreas That Is Metastatic or Cannot Be Removed by Surgery. 3 November 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02595424?term=ECOG-ACRIN+EA2142&rank=1 (accessed on 30 May 2019).


{kind=link}
| Guidelines Situations | Canadian Consensus Guidelines 2016 (12) | NANETS (North American Neuroendocrine Tumor Society) Guidelines 2017 (13–14) | NCCN (National Comprehensive Cancer Network) Guidelines 2018 (15) | ENETS (European Neuroendocrine Tumor Society) Guidelines 2016 (16–17) |
|---|---|---|---|---|
| Locoregional disease | → Complete resection of the primary tumor and the associated lymphatic drainage field → Evaluate for multifocality (Unclear if favor open vs. laparoscopic minimally invasive) | → Complete open resection of primary tumor and the associated lymphatic drainage field → Laparoscopic resection is acceptable if bowel can be completely run through a small incision | → Complete resection of the primary tumor and the associated lymphatic drainage field → Evaluate for multifocality (Unclear if favor open vs. laparoscopic/ minimally invasive) | → Complete open (or in selected patient laparoscopic) resection of primary tumor and the associated lymphatic drainage field |
| Residual disease or positive margins post-primary resection | → Definitive resection when technically feasible | → Not addressed | → Not addressed | → Not addressed |
| Synchronous primary and metastatic disease | → Resection of primary tumor(s), lymph nodes in combination with liver metastases | → Resection of primary tumor(s), lymph nodes in combination with liver metastases | → Resection of primary tumor(s), lymph nodes in combination with liver metastases | → Local radical open resection of primary tumor(s), lymph nodes in combination with liver metastases |
| Liver metastases | → Resection of liver metastases with the goal of preserving liver parenchyma and both left and right inflow and outflow vascular patency when possible. → Image-guided ablation either alone for limited disease (tumors ideally < 3 cm) or in combination with surgery → If cytoreductive surgical/ablative procedures not indicated; bland embolization, chemoembolization or radioembolization | → Resection of liver metastases should be attempted when feasible and low morbidity/mortality → Parenchymal sparing procedures should be considered → Patients with any number or size of metastases, intermediate grade, extrahepatic disease should be considered for liver debulking operations if a 70% debulking threshold can be achieved. → If cytoreductive surgery not indicated; bland embolization, chemoembolization or radioembolization | → Resection of liver metastases or ablative therapies such as RFA (Radio-Frequency Ablation) or cryoablation may be considered if near-complete treatment of tumor burden can be achieved. → If cytoreductive surgical/ablative procedures not indicated; bland embolization, chemoembolization or radioembolization | → Resection of liver metastases when feasible → Image-guided ablation can be combined with surgical resection → If cytoreductive surgical procedures not indicated: radio-frequency ablation, laser-induced thermotherapy, transarterial chemoembolization, transarterial embolization or selective internal radiation therapy (investigational) |
| Peritoneal metastases | → Surgical resection when feasible and synchronous resection of peritoneal disease and hepatic metastasectomy is an option. | → Surgical resection when feasible → No evidence supporting use of HIPEC (hyperthermic intraperitoneal chemotherapy) | → Not addressed | → Not addressed |
| Abdominal disease in setting of extra-abdominal metastases | → Cytoreduction should be considered in selected patients for symptom control | → Not addressed | → Not addressed | → Not addressed |
| Prophylactic cholecystectomy | → Consider as part of any abdominal surgical procedure | → If future treatment with SSA is anticipated, a prophylactic cholecystectomy can be considered | → If future treatment with SSA is anticipated, a prophylactic cholecystectomy can be considered | → If future treatment with SSA is anticipated, a prophylactic cholecystectomy can be considered |
| Systemic therapy for metastatic or unresectable disease | → First line: somatostatin analogues (octreotide LAR (Long Acting Release), lanreotide autogel) → Second line: everolimus single agent OR everolimus + SSA OR PRRT (Peptide Radionuclide Radiation Therapy) | → First line: somatostatin analogues (octreotide LAR, lanreotide autogel) → Second line: PRRT (preferred) OR everolimus → Other less favored modalities: Interferon alpha 2b or locoregional therapy | → First line: somatostatin analogues (octreotide LAR, lanreotide autogel) or watch and wait if asymptomatic with low tumor burden → Second line: start SSA if prior watch and wait OR everolimus OR PRRT OR locoregional therapy OR interferon alpha 2b OR cytotoxic chemotherapy if no other options feasible | → First line: somatostatin analogues (octreotide LAR, lanreotide autogel) or watch and wait if G1, low tumor burden, stable and asymptomatic → Second line: Start SSA if prior watch and wait OR locoregional therapy OR PRRT OR everolimus OR interferon alpha 2b |
| Symptom control—Carcinoid syndrome | → First line: somatostatin analogues (octreotide LAR, lanreotide Autogel) → Short-acting octreotide recommended for two weeks after first long-acting SSA injection → Refractory to SSA: telotristat etiprate OR interferon alpha OR SSA dose escalation | → First line: somatostatin analogues (octreotide LAR, lanreotide autogel) → Short-acting octreotide recommended for two weeks after first long-acting SSA injection → Refractory to SSA: telotristate etiprate | → First line: Somatostatin analogues (Octreotide LAR, Lanreotide Autogel) → Short-acting octreotide recommended for two weeks after first long-acting SSA injection → Refractory to SSA: Telotristate etiprate | → First line: somatostatin analogues (octreotide LAR, lanreotide autogel) → Refractory to SSA: SSA dose increase OR add-on interferon alpha 2b OR pasireotide OR PRRT OR telotristat etiprate |
| Symptom control—Bone and brain metastases | → External beam radiotherapy is an option | → Not addressed | → Not addressed | → Not addressed |
| Trial | Eligibility Criteria | Intervention | n | Median PFS | Adverse Effects | Health-Related Quality of Life |
|---|---|---|---|---|---|---|
| PROMID (8) 2009 | Treatment-naïve midgut NETs, secretory/NS | Octreotide LAR 30 mg IM q4w vs. Placebo IM q4w | 85 (42 vs. 43) | Median Time-to-Tumor-Progression 14.3 mo vs. 6.0 mo HR = 0.34 (0.2–0.59), p 0.000072 | Any AES: 26 vs. 23 Hematological AES: 12 vs. 2 Fatigue and fever: 19 vs. 5 | PROMID HRQoL Study (48) Time to definitive deterioration (OC vs. PBO) - 18.5 mo vs. 6.8 mo (fatigue) - NR vs. 18.2 mo (pain) - NR vs. 16.4 mo (insomnia) NR: Not reached |
| CLARINET (9) 2014 | Previous treatment permitted, enteropancreatic NETs, non-secretory, SSR+ | Lanreotide Autogel 120 mg dsq q4w vs. Placebo sc q4w | 204 (101 vs. 103) | NR vs. 18 mo HR = 0.47 (0.30–0.73), p < 0.001 | Diarrhea: 26 vs. 9 Abdominal pain: 14 vs. 2 Any AES: 25 vs. 31 | No HRQoL study |
| RADIANT-4 (10) 2016 | Advanced lung or GI NETs, nonsecreting, prior treatment allowed | Everolimus 10 mg po die vs. Placebo | 302 (205 vs. 97) | 11 vs. 3.9 mo HR 0.48 (0.35–0.67), p < 0.00001 | Stomatitis: 63 vs. 19 Diarrhea: 31 vs. 16 Infections: 29 vs. 4 Rash: 27 vs. 8 Peripheral edema: 26 vs. 4 Anemia: 16 vs. 2 Anorexia: 16 vs. 6 Asthenia: 16 vs. 5 Pneumonitis: 16 vs. 1 Dysgueusia: 15 vs. 4 Cough: 13 vs. 3 Stomatitis: 8 vs. 0 Diarrhea: 7 vs. 2 | RADIANT-4 HRQoL Study (49) Time to definitive deterioration (FACT-G global score) (EV vs. PBO) 11.27 mo vs. 9.23 mo (AHR 0.81, p 0.31) |
| NETTER-1 (11) 2015 | NETs progressing on Octreotide LAR (30 mg), midgut NETs, secretory1nonsecretory, SSR + | 7.4 GBq 177-Lu-Dotatate q8w + Octreotide LAR 30 mg IM q4w vs. Octreotide LAR 60 mg IM q4w | 229 (116 vs. 113) | NR vs. 8.4 mo HR = 0.209 (0.129–0.388), p < 0.0001 | Any AES: 86 vs. 31 | NETTER-1 HRQoL Study (50) Time to definitive deterioration (PRRT vs. PBO) Longer - Global health status 28.8 mo vs. 6.1 mo (HR 0.406) - Physical functioning 25.2 vs. 11.5 mo (HR 0.518) - Role functioning (HR 0.580) - Fatigue (HR 0.621) - Pain (HR 0.566) - Diarrhea (HR 0.473) - Disease-related worries (HR 0.572) Body image (HR 0.425) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larouche, V.; Akirov, A.; Alshehri, S.; Ezzat, S. Management of Small Bowel Neuroendocrine Tumors. Cancers 2019, 11, 1395. https://doi.org/10.3390/cancers11091395
Larouche V, Akirov A, Alshehri S, Ezzat S. Management of Small Bowel Neuroendocrine Tumors. Cancers. 2019; 11(9):1395. https://doi.org/10.3390/cancers11091395
Chicago/Turabian StyleLarouche, Vincent, Amit Akirov, Sameerah Alshehri, and Shereen Ezzat. 2019. "Management of Small Bowel Neuroendocrine Tumors" Cancers 11, no. 9: 1395. https://doi.org/10.3390/cancers11091395