Repurposing Penfluridol in Combination with Temozolomide for the Treatment of Glioblastoma

 ,
,
Abstract
:1. Introduction
2. Results
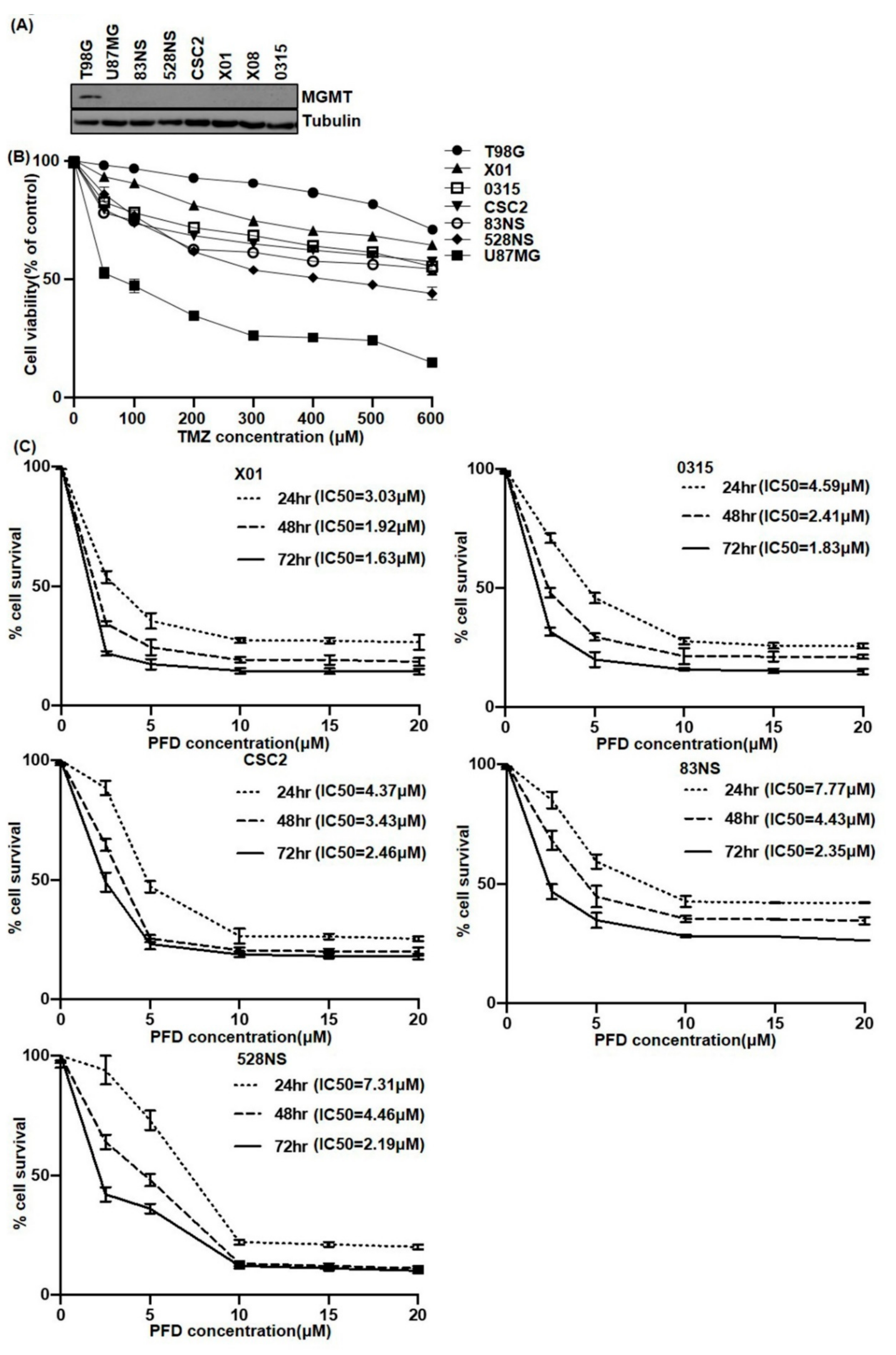
2.1. The Effect of PFD on GSCs Proliferation
2.2. PFD Suppresses the Stemness of GSCs
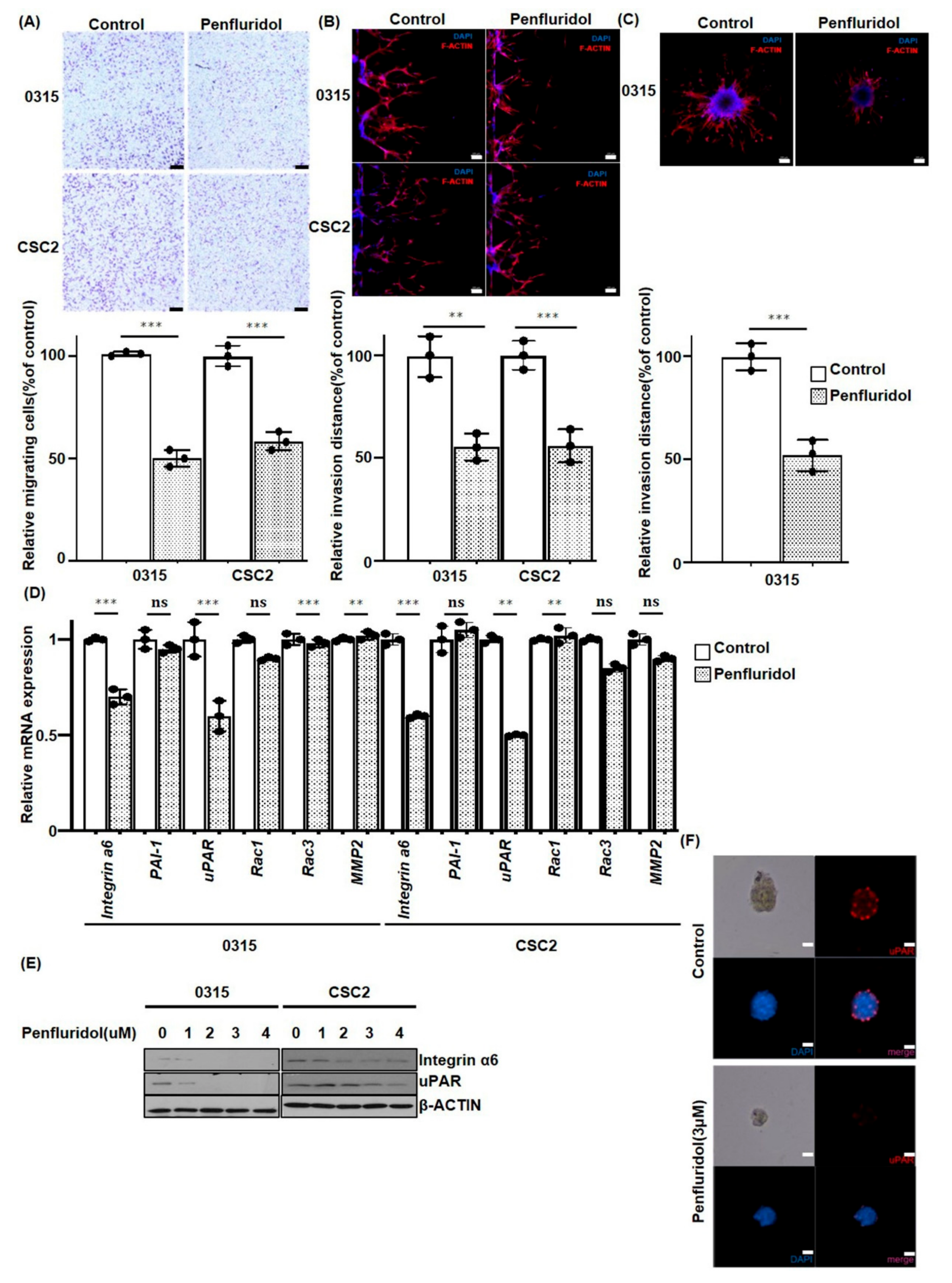
2.3. PFD Inhibits the Migration and Invasion of GSCs
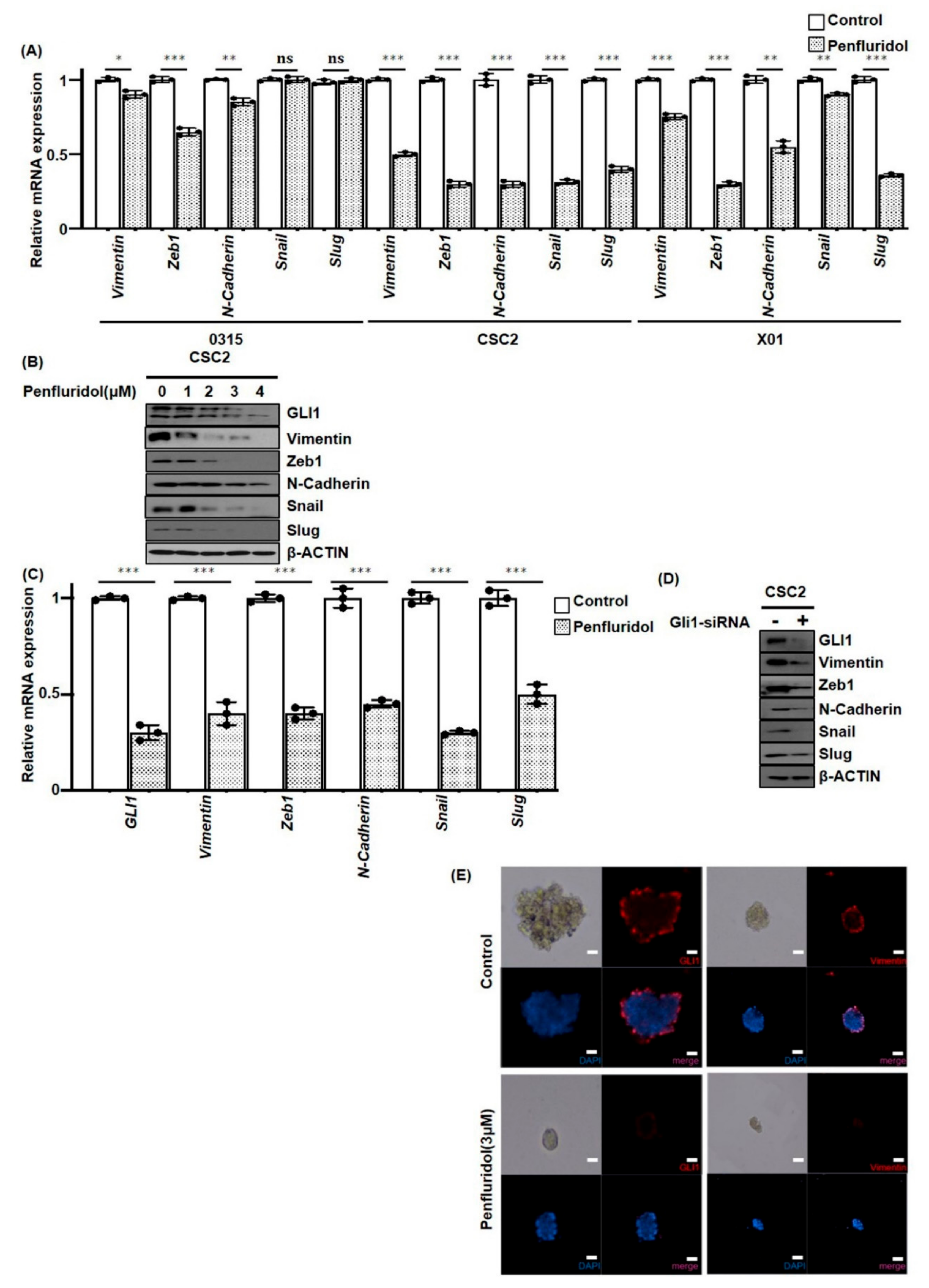
2.4. PFD Suppresses Epithelial-Mesenchymal Transition Induced by GLI1 in GSCs
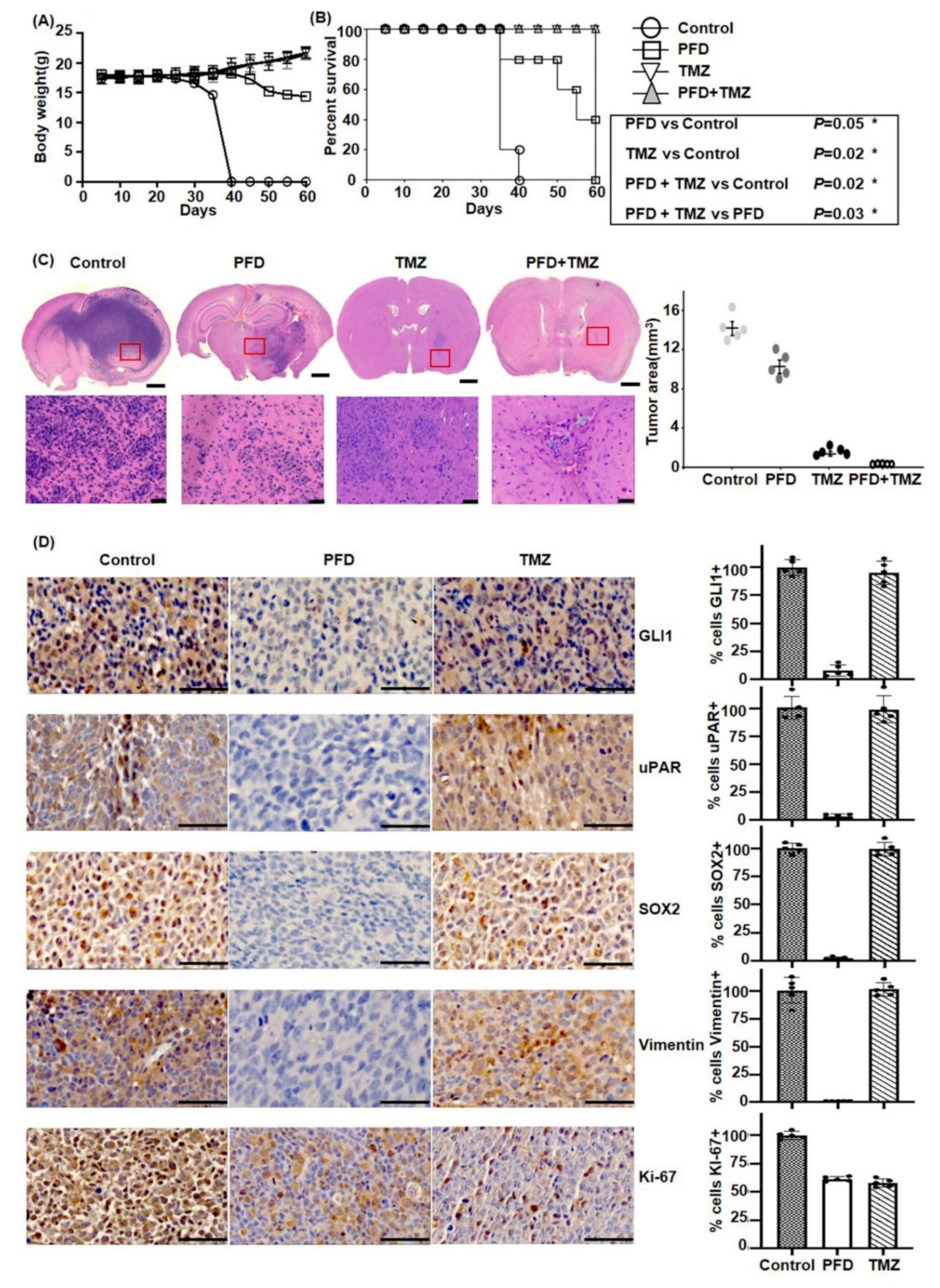
2.5. PFD in Combination with TMZ Significantly Reduces GBM Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Drugs Treatment
4.2. Immunoblotting and Antibodies
4.3. Reverse Transcription and Quantitative PCR (RT-qPCR)
4.4. Primer
4.5. Migration and Invasion Assay
4.6. Invasion Assay in the Microfluidic Device
4.7. Sphere Formation Assay
4.8. GLI1 Silencing
4.9. Immunocytochemistry
4.10. In Vivo Tumor Models
- Group 1 (vehicle): daily oral administration of normal saline;
- Group 2: daily oral administration of PFD, 0.8 mg/kg/week from day 3 to 30;
- Group 3: oral administration of TMZ, 33 mg/kg/5 days/week, 3 cycles from day 3 to 24;
- Group 4: TMZ and PFD combination for up to day 30.
4.11. Immunohistochemistry
4.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol. 2015, 17 (Suppl. 4), iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.M.; Kang, S.-H.; Park, C.-K.; Jung, S.; Park, E.S.; Lee, J.-S.; Kim, S.-H.; Woo, H.G. Recurrent glioblastomas reveal molecular subtypes associated with mechanistic implications of drug-resistance. PLoS ONE 2015, 10, e0140528. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Kurz, S.C.; Cabrera, L.P.; Hastie, D.; Huang, R.; Unadkat, P.; Rinne, M.; Nayak, L.; Lee, E.Q.; Reardon, D.A.; Wen, P.Y. PD-1 inhibition has only limited clinical benefit in patients with recurrent high-grade glioma. Neurology 2018, 91, e1355–e1359. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, F.H.-C.; Tsai, K.-Y.; Su, C.-Y.; Lee, C.-C. The incidence and relative risk factors for developing cancer among patients with schizophrenia: A nine-year follow-up study. Schizophr. Res. 2011, 129, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Tabarés-Seisdedos, R.; Rubenstein, J.L.R.; Tabar, R. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: Implications for schizophrenia, autism and cancer. Mol. Psychiatry 2009, 14, 563–589. [Google Scholar] [CrossRef] [PubMed]
- Yovel, G.; Sirota, P.; Mazeh, D.; Shakhar, G.; Rosenne, E.; Ben-Eliyahu, S. Higher Natural Killer Cell Activity in Schizophrenic Patients: The Impact of Serum Factors, Medication, and Smoking. Brain Behav. Immun. 2000, 14, 153–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiklund, E.D.; Catts, V.S.; Catts, S.V.; Ng, T.F.; Whitaker, N.J.; Brown, A.J.; Lutze-Mann, L.H.; Lutze-Mann, L.H. Cytotoxic effects of antipsychotic drugs implicate cholesterol homeostasis as a novel chemotherapeutic target. Int. J. Cancer 2010, 126, 28–40. [Google Scholar] [CrossRef]
- Kang, S.; Hong, J.; Lee, J.M.; Moon, H.E.; Jeon, B.; Choi, J.; Yoon, N.A.; Paek, S.H.; Roh, E.J.; Lee, C.J.; et al. Trifluoperazine, a well-known antipsychotic, inhibits glioblastoma invasion by binding to calmodulin and disinhibiting calcium release channel IP3R. Mol. Cancer Ther. 2017, 16, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Soares, B.G.; De Lima, M.S. Penfluridol for schizophrenia. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef]
- Wu, L.; Liu, Y.-Y.; Li, Z.-X.; Zhao, Q.; Wang, X.; Yu, Y.; Wang, Y.-Y.; Wang, Y.-Q.; Luo, F. Anti-tumor effects of penfluridol through dysregulation of cholesterol homeostasis. Asian Pac. J. Cancer Prev. 2014, 15, 489–494. [Google Scholar] [CrossRef]
- Ranjan, A.; Gupta, P.; Srivastava, S.K. Penfluridol: An antipsychotic agent suppresses metastatic tumor growth in triple-negative breast cancer by inhibiting integrin signaling axis. Cancer Res. 2016, 76, 877–890. [Google Scholar] [CrossRef]
- Ranjan, A.; Srivastava, S.K. Penfluridol suppresses glioblastoma tumor growth by Akt-mediated inhibition of GLI1. Oncotarget 2017, 8, 32960–32976. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Park, G.; Kim, T.H.; Hong, J.H.; Kim, Y.J.; Jin, X.; Kang, S.; Jung, J.E.; Kim, J.Y.; Yun, H.; et al. Pigment epithelium-derived factor (PEDF) expression induced by egfrviii promotes self-renewal and tumor progression of glioma stem cells. PLoS Biol. 2015, 13, e1002152. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; MacSwords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Clément, V.; Sánchez, P.; De Tribolet, N.; Radovanovic, I.; Altaba, A.R.I. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Gilder, A.S.; Natali, L.; Van Dyk, D.M.; Zalfa, C.; Banki, M.A.; Pizzo, D.P.; Wang, H.; Klemke, R.L.; Mantuano, E.; Gonias, S.L. The urokinase receptor induces a mesenchymal gene expression signature in glioblastoma cells and promotes tumor cell survival in neurospheres. Sci. Rep. 2018, 8, 2982. [Google Scholar] [CrossRef] [PubMed]
- Raeder, M.B.; Fernø, J.; Vik-Mo, A.O.; Steen, V.M. SREBP activation by antipsychotic- and antidepressant-drugs in cultured human liver cells: relevance for metabolic side-effects? Mol. Cell. Biochem. 2006, 289, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Burattini, L.; Nabissi, M.; Morelli, M.B.; Berardi, R.; Santoni, G.; Cascinu, S. Essential role of Gli proteins in glioblastoma multiforme. Curr. Protein Pept. Sci. 2013, 14, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, J.; Zhao, S.; Yao, K.; Sun, Y.; Li, Y.; Chen, L.; Li, R.; Zhai, X.; Zhang, J.; et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 184. [Google Scholar] [CrossRef]
- Cheng, H.-W.; Liang, Y.-H.; Kuo, Y.-L.; Chuu, C.-P.; Lin, C.-Y.; Lee, M.-H.; Wu, A.T.H.; Yeh, C.-T.; Chen, E.I.-T.; Whang-Peng, J.; et al. Identification of thioridazine, an antipsychotic drug, as an antiglioblastoma and anticancer stem cell agent using public gene expression data. Cell Death Dis. 2015, 6, e1753. [Google Scholar] [CrossRef]
- Riaz, S.K.; Ke, Y.; Wang, F.; Kayani, M.A.; Malik, M.F.A. Influence of SHH/GLI1 axis on EMT mediated migration and invasion of breast cancer cells. Sci. Rep. 2019, 9, 6620. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Wright, S.; Srivastava, S.K. Immune consequences of penfluridol treatment associated with inhibition of glioblastoma tumor growth. Oncotarget 2017, 8, 47632–47641. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Rohnalter, V.; Bergström, Å.; Kooshesh, M.; Svenningsson, P.; Toftgård, R. Antipsychotic drugs regulate hedgehog signaling by modulation of 7-Dehydrocholesterol reductase levels. Mol. Pharmacol. 2010, 78, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Nikvarz, N.; Vahedian, M.; Khalili, N. Chlorpromazine versus penfluridol for schizophrenia. Cochrane Database Syst. Rev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.R.; Ommurugan, B.; Adiga, S.; Patil, N.; Reddy, S. Penfluridol induced extrapyramidal symptoms (EPS): A case report. J. Appl. Pharm. Sci. 2017, 7, 214–216. [Google Scholar] [CrossRef]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Moreno-Cugnon, L.; Barrena, C.; Villanua, J.; Ruiz, I.; et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Han, S.; Jeon, J.S.; Yamamoto, K.; Zervantonakis, I.K.; Sudo, R.; Kamm, R.D.; Chung, S. Microfluidic assay for simultaneous culture of multiple cell types on surfaces or within hydrogels. Nat. Protoc. 2012, 7, 1247–1259. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Piao, Y.; Park, S.Y.; Henry, V.; Smith, B.D.; Tiao, N.; Flynn, D.L.; De Groot, J.F. Novel MET/TIE2/VEGFR2 inhibitor altiratinib inhibits tumor growth and invasiveness in bevacizumab-resistant glioblastoma mouse models. Neuro Oncol. 2016, 18, 1230–1241. [Google Scholar] [CrossRef] [Green Version]
- Shu, J.; Dolman, G.E.; Duan, J.; Qiu, G.; Ilyas, M. Statistical colour models: An automated digital image analysis method for quantification of histological biomarkers. Biomed. Eng. Online 2016, 15, 46. [Google Scholar] [CrossRef]
- Kumar, R.M.R.; Fuchs, B. Hedgehog signaling inhibitors as anti-cancer agents in osteosarcoma. Cancers 2015, 7, 784–794. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SOX2 | F | AGAAGAGGAGAGAGAAAGAAAGGGAGAGA |
| R | GAGAGAGGCAAACTGGAATCAGGATCAAA | |
| NESTIN | F | CAGCGTTGGAACAGAGGTTGG |
| R | TGGCACAGGTGTCTCAAGGGTAG | |
| OCT4 | F | AACCTGGAGTTTGTGCCAGGGTTT |
| R | TGAACTTCACCTTCCCTCCAACCA | |
| Integrin-a6 | F | GTGGCTATTCTCGCTGGGAT |
| R | ACCTAGAGCGTTTAAAGAATCCAC | |
| PAI-1 | F | ACAAGTTCAACTATACTGAGTTCACCACGCCC |
| R | TGAAACTGTCTGAACATGTCGGTCATTCCC | |
| uPAR | F | CCTCTGCAGGACCACGAT |
| R | TGGTCTTCTCTGAGTGGGTAC | |
| Rac1 | F | CTGAAGGAGAAGAAGCTGAC |
| R | TCGTCAAACACTGTCTTGAG | |
| Rac3 | F | GACGACAAGGACACCATTGA |
| R | CCTCGTCAAACACTGTCTTC | |
| MMP2 | F | GATAACCTGGATGCCGTCGT |
| R | CGAAGGCAGTGGAGAGGAAG | |
| GLI1 | F | CAACTCGATGACCCCACCAC |
| R | CAGACAGTCCTTCTGTCCCCA | |
| Vimentin | F | CCAGGCAAAGCAGGAGTC |
| R | CGAAGGTGACGAGCCATT | |
| Zeb1 | F | GTGGCGGTAGATGGTAAT |
| R | CTGTTTGTAGCGACTGGA | |
| N-Cadherin | F | TGCAAGACTGGATTTCCTGAAGA |
| R | AGCTTCTCACGGCATACACC | |
| Snail | F | CGGAAGCCTAACTACAGCGA |
| R | GCCAGGACAGAGTCCCAGAT | |
| Slug | F | AGATGCATATTCGGACCCAC |
| R | CCTCATGTTTGTGCAGGAGA | |
| HPRT1 | F | AAAGGACCCCACGAAGTGTT |
| R | AAGCAGATGGCCACAGAACTA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Chong, K.; Ryu, B.-K.; Park, K.-J.; Yu, M.O.; Lee, J.; Chung, S.; Choi, S.; Park, M.-J.; Chung, Y.-G.; et al. Repurposing Penfluridol in Combination with Temozolomide for the Treatment of Glioblastoma. Cancers 2019, 11, 1310. https://doi.org/10.3390/cancers11091310
Kim H, Chong K, Ryu B-K, Park K-J, Yu MO, Lee J, Chung S, Choi S, Park M-J, Chung Y-G, et al. Repurposing Penfluridol in Combination with Temozolomide for the Treatment of Glioblastoma. Cancers. 2019; 11(9):1310. https://doi.org/10.3390/cancers11091310
Chicago/Turabian StyleKim, Hyungsin, Kyuha Chong, Byung-Kyu Ryu, Kyung-Jae Park, Mi OK Yu, Jihye Lee, Seok Chung, Seongkyun Choi, Myung-Jin Park, Yong-Gu Chung, and et al. 2019. "Repurposing Penfluridol in Combination with Temozolomide for the Treatment of Glioblastoma" Cancers 11, no. 9: 1310. https://doi.org/10.3390/cancers11091310