Proteomic Analysis of Breast Cancer Resistance to the Anticancer Drug RH1 Reveals the Importance of Cancer Stem Cells


 , and
, and
Abstract
:
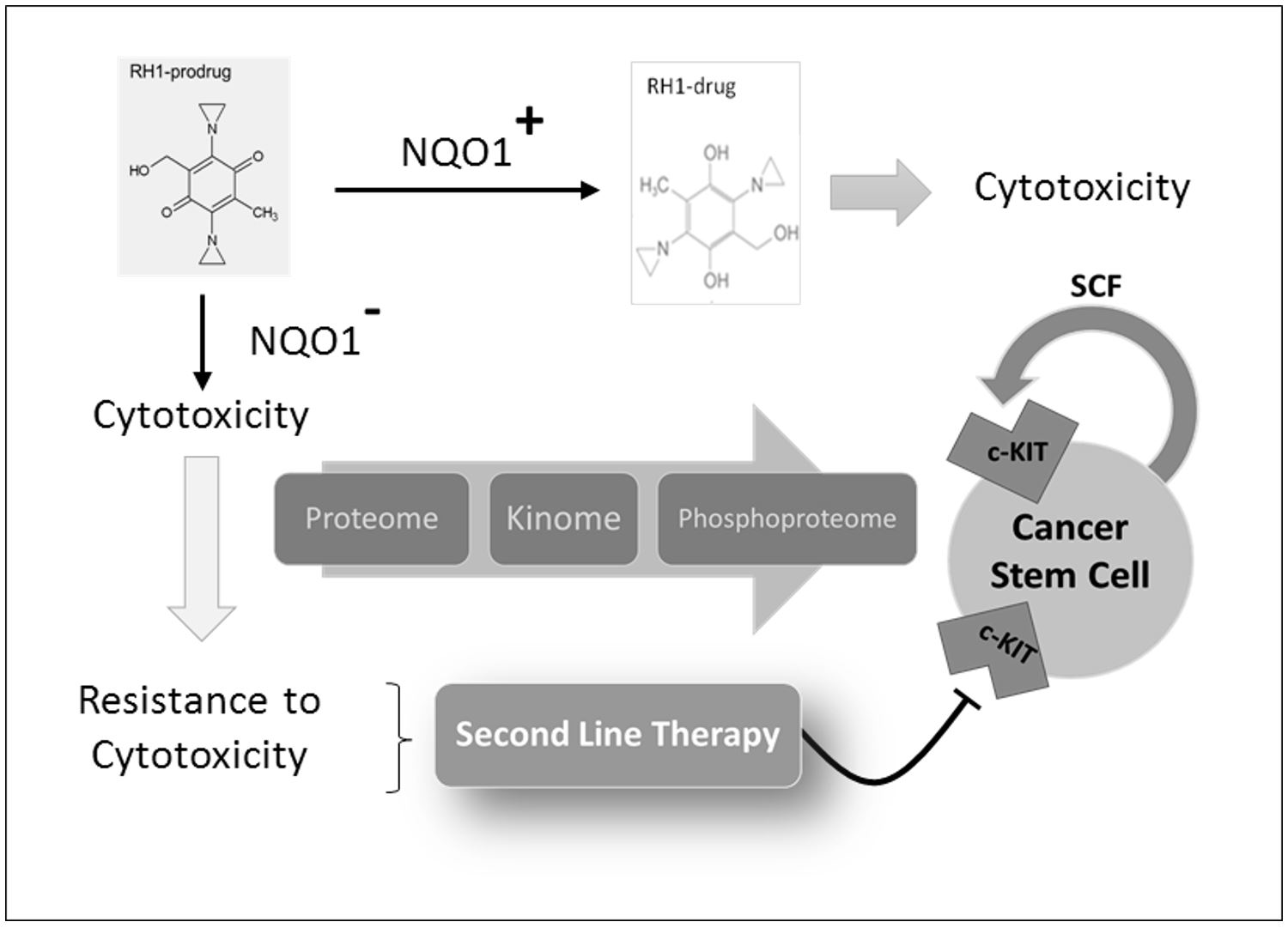
1. Introduction
2. Results and Discussion
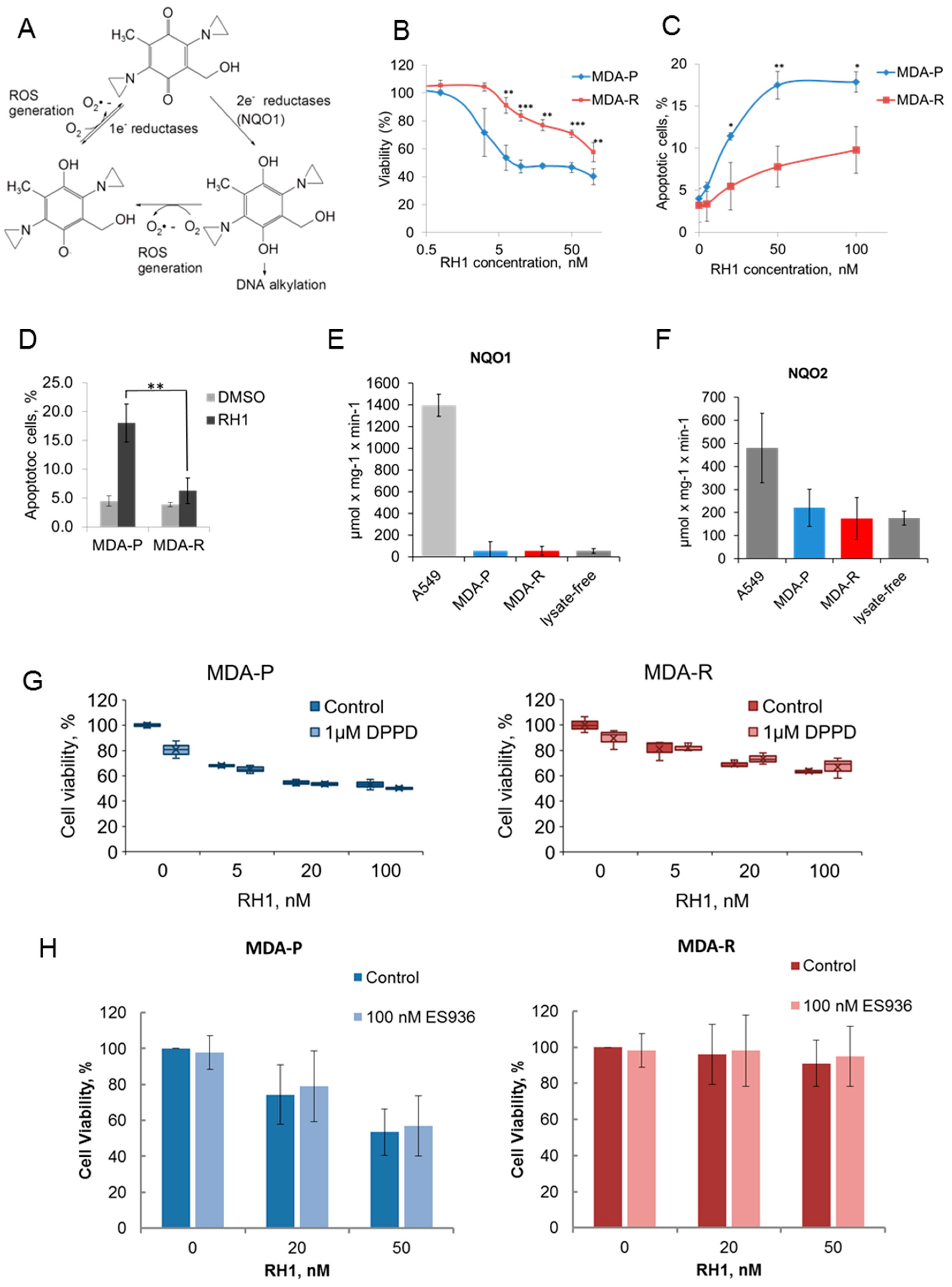
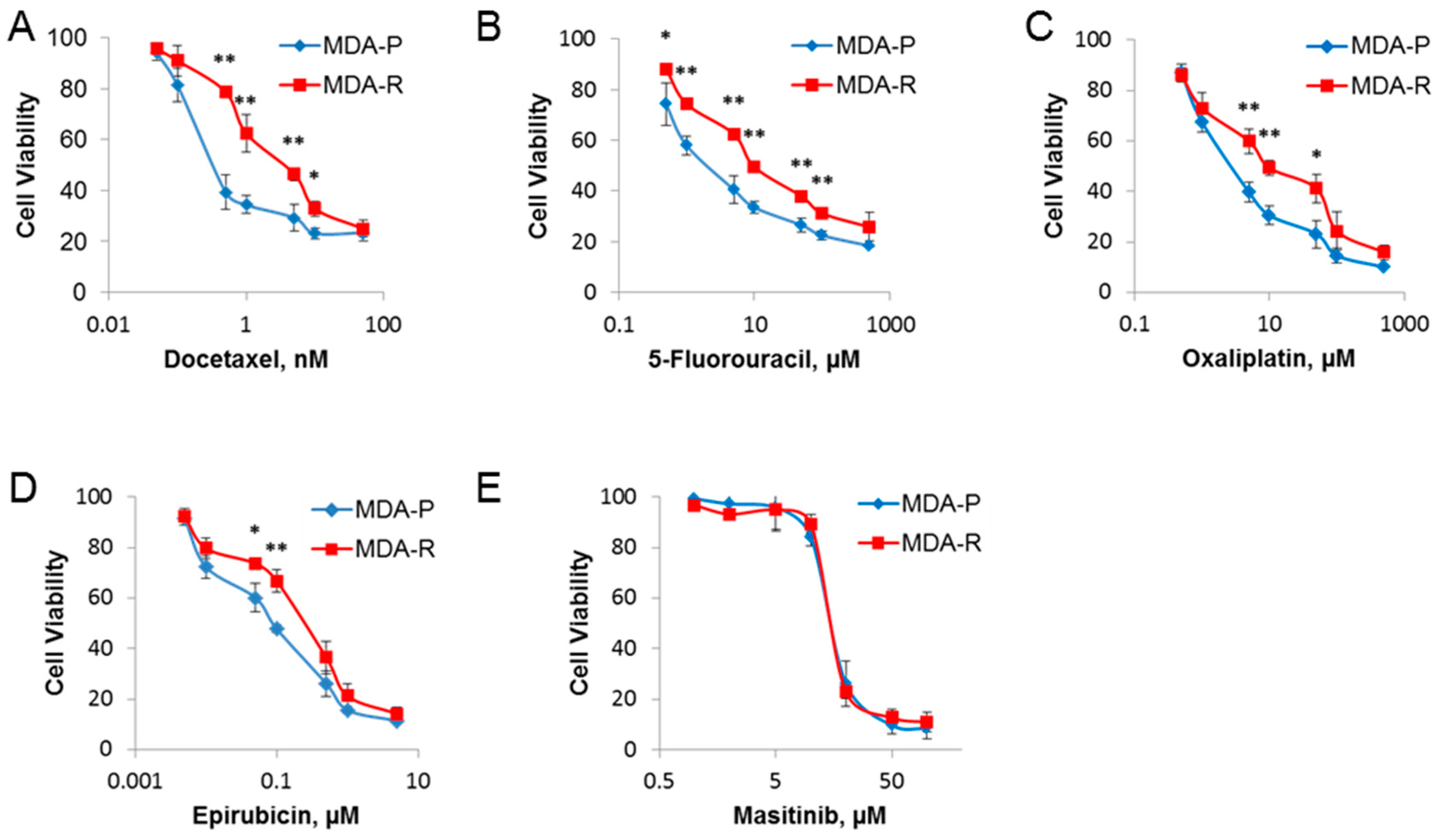
2.1. Establishment and Characterization of RH1-Resistant MDA-MB-231 Cells
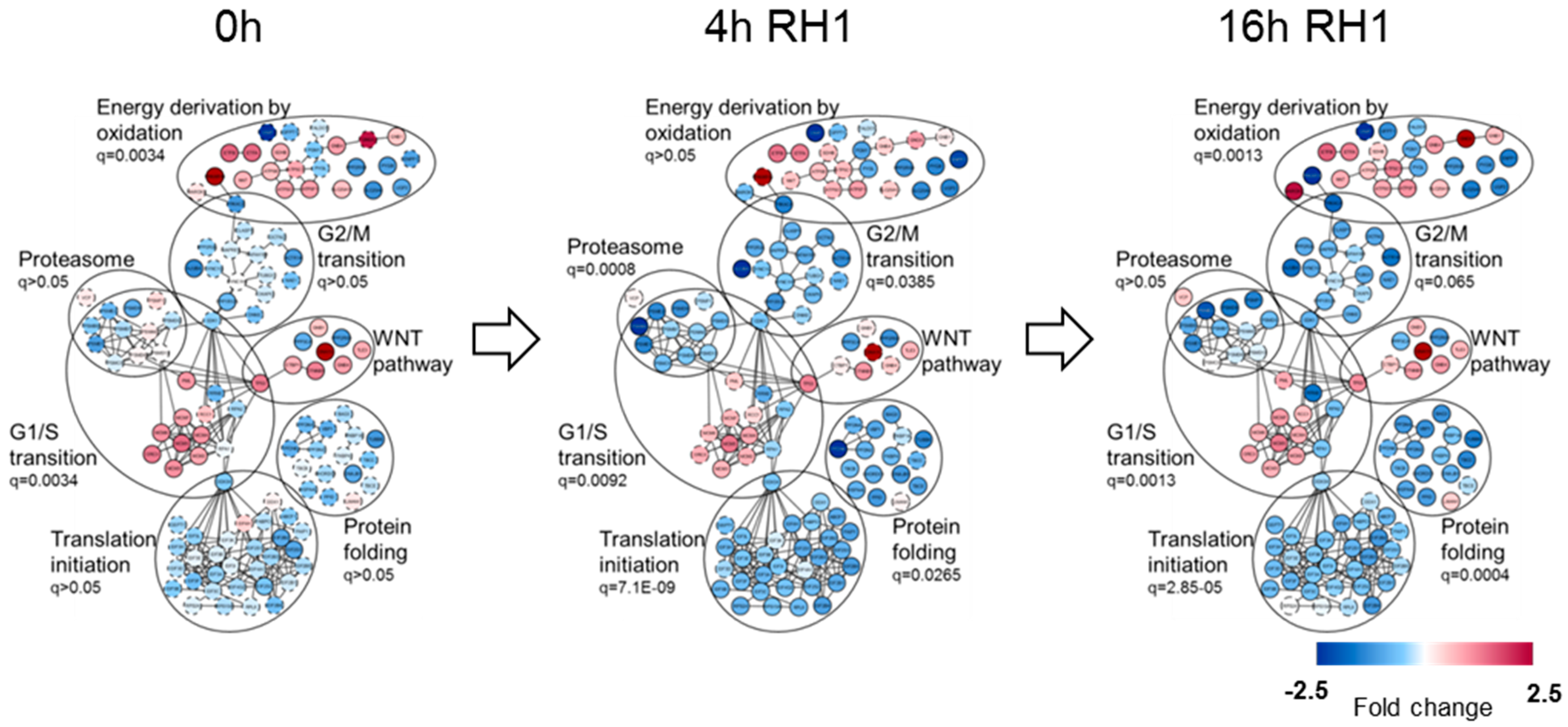
2.2. Differential Global Proteomics of Breast Cancer Cells Predicts Cellular Mechanisms of RH1 Resistance
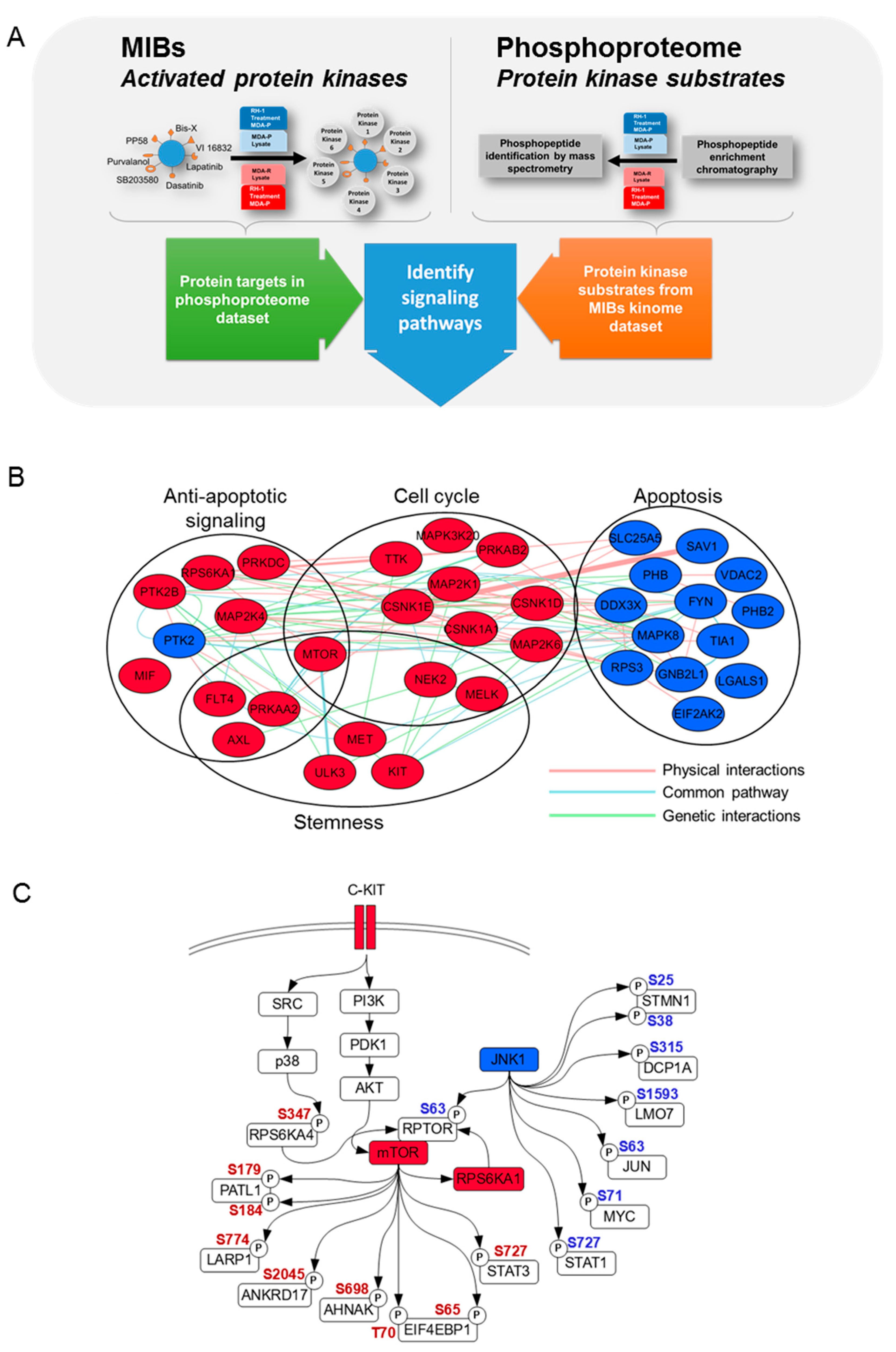
2.3. Phosphoproteome and Kinome of RH1 Resistant Breast Cancer Cells Highlight Signaling Pathways Responsible for the RH1 Resistance
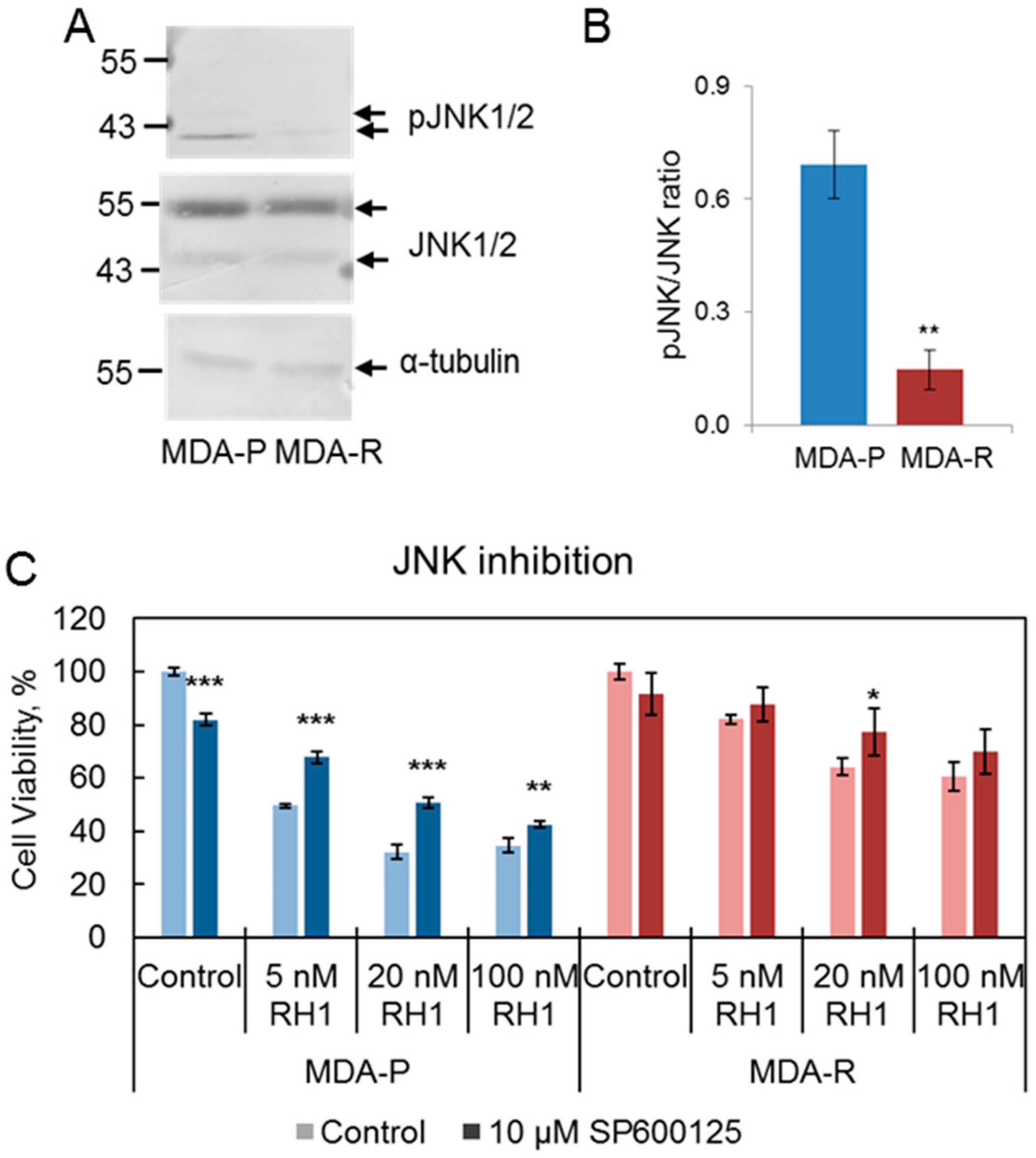
2.4. Decrease in JNK Activity Mediates Resistance to RH1
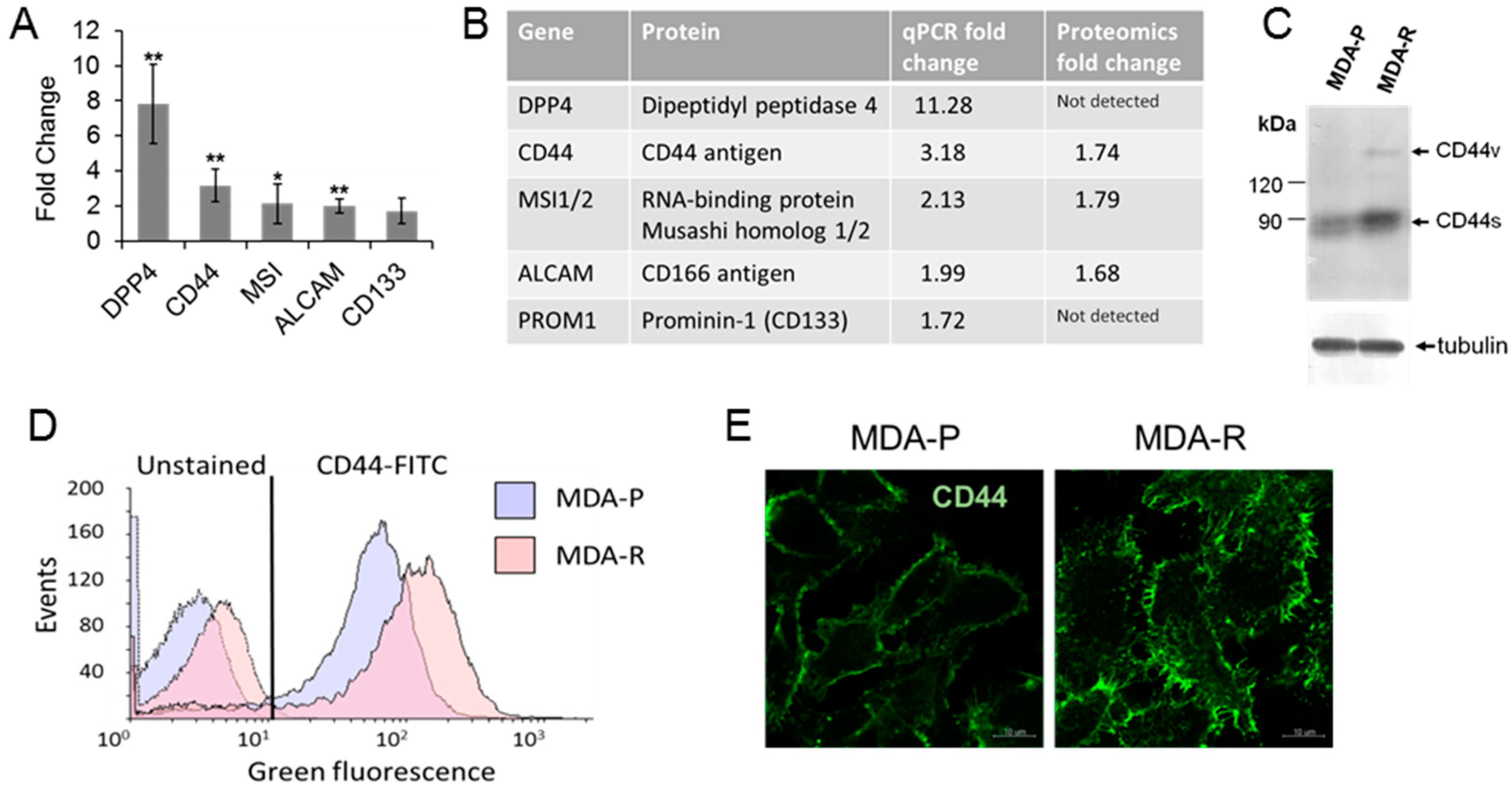
2.5. Cancer Stem Cell Population Is Enriched in the Breast Cancer Cells Resistant to RH1
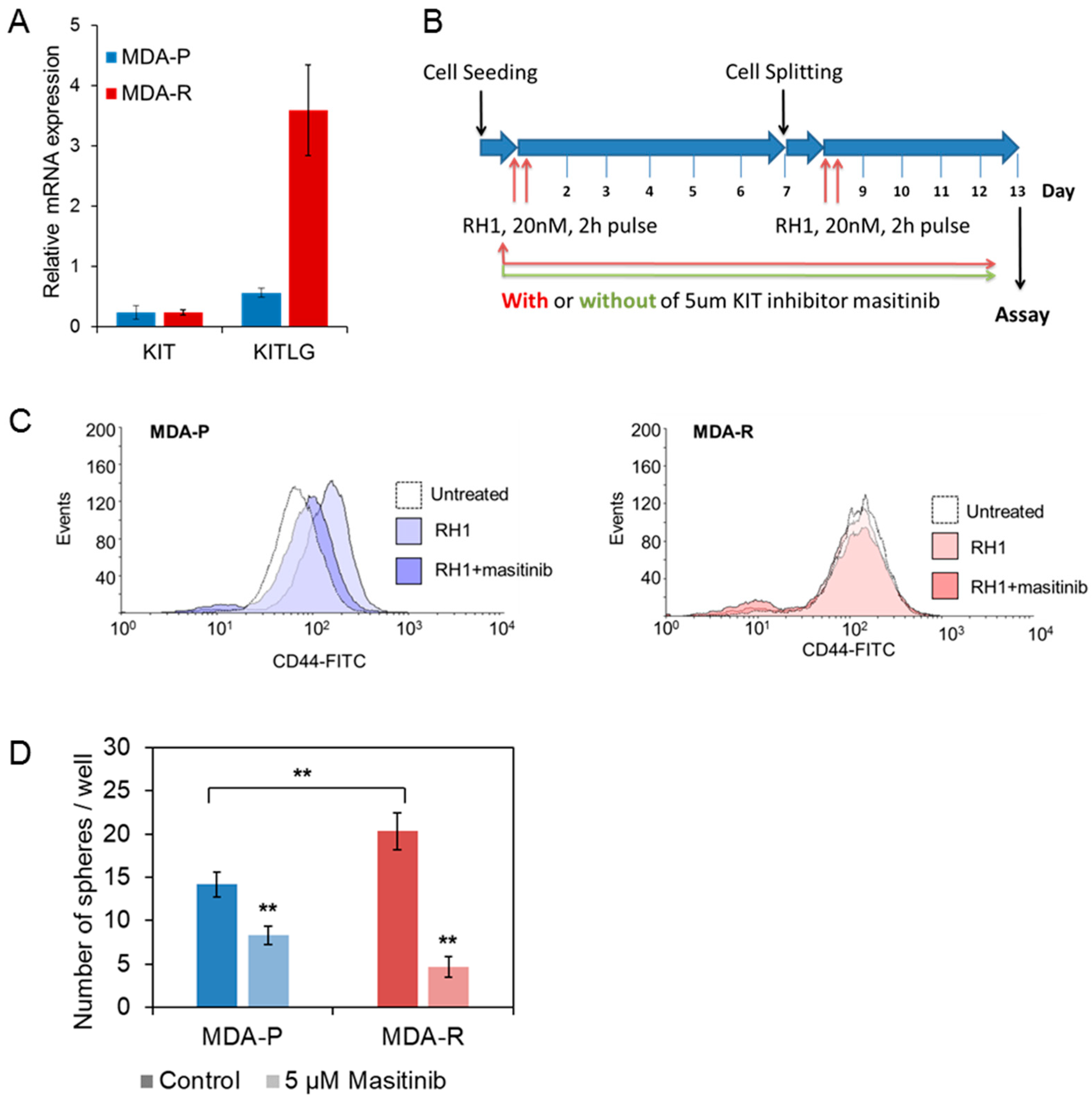
2.6. Stem Cell Factor Receptor c-KIT Contributes to the Acquisition of RH1 Resistance in Breast Cancer Cells
2.7. c-KIT Is a Potential Target in the Therapy of RH1-Resistant Breast Cancer Cells
3. Materials and Methods
3.1. Cell Culture, Drug Treatment and Establishment of RH1-Resistant Cell Lines
3.2. Antibodies and Inhibitors
3.3. Assessment of Cell Viability and Apoptosis
3.4. RH1 Reduction Assay
3.5. Flow Cytometry and Western Blot
3.6. Sphere-Forming Assay
3.7. Total RNA Isolation and Real-Time qPCR for Evaluation of the mRNA Expression
3.8. Cell lysis and Sample Preparation for Mass Spectrometry Analysis
3.9. Multiplexed Inhibitor Bead Affinity Extraction
3.10. Liquid Chromatography and Mass Spectrometry
3.11. Data Processing, Searching and Analysis
3.12. Computational Functional Analysis of Proteomic Data
3.13. Phosphoproteome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Abubaker, K.; Latifi, A.; Luwor, R.; Nazaretian, S.; Zhu, H.; Quinn, M.A.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer 2013, 12, 24. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Lavi, O.; Hall, M.D.; Gillet, J.-P. Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 85–102. [Google Scholar] [CrossRef]
- Yeo, S.K.; Guan, J.-L. Hierarchical heterogeneity in mammary tumors and its regulation by autophagy. Autophagy 2016, 12, 1960–1961. [Google Scholar] [CrossRef]
- Hu, Y.; Fu, L. Targeting cancer stem cells: A new therapy to cure cancer patients. Am. J. Cancer Res. 2012, 2, 340–356. [Google Scholar]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Shigdar, S.; Gantier, M.P.; Hou, Y.; Wang, L.; Li, Y.; Al Shamaileh, H.; Yin, W.; Zhou, S.-F.; Zhao, X.; et al. Cancer stem cell targeted therapy: progress amid controversies. Oncotarget 2015, 6, 44191–44206. [Google Scholar] [CrossRef]
- Yang, T.; Rycaj, K. Targeted therapy against cancer stem cells. Oncol. Lett. 2015, 10, 27–33. [Google Scholar] [CrossRef]
- Danson, S.J.; Johnson, P.; Ward, T.H.; Dawson, M.; Denneny, O.; Dickinson, G.; Aarons, L.; Watson, A.; Jowle, D.; Cummings, J.; et al. Phase I pharmacokinetic and pharmacodynamic study of the bioreductive drug RH1. Ann. Oncol. 2011, 22, 1653–1660. [Google Scholar] [CrossRef]
- Yan, C.; Kepa, J.K.; Siegel, D.; Stratford, I.J.; Ross, D. Dissecting the Role of Multiple Reductases in Bioactivation and Cytotoxicity of the Antitumor Agent 2,5-Diaziridinyl-3-(hydroxymethyl)-6-methyl-1,4-benzoquinone (RH1). Mol. Pharmacol. 2008, 74, 1657–1665. [Google Scholar] [CrossRef] [Green Version]
- Tudor, G.; Alley, M.; Nelson, C.M.; Huang, R.; Covell, D.G.; Gutierrez, P.; Sausville, E.A. Cytotoxicity of RH1: NAD(P)H:quinone acceptor oxidoreductase (NQO1)-independent oxidative stress and apoptosis induction. Anticancer Drugs 2005, 16, 381–391. [Google Scholar] [CrossRef]
- Dehn, D.L.; Inayat-Hussain, S.H.; Ross, D. RH1 induces cellular damage in an NAD(P)H:quinone oxidoreductase 1-dependent manner: Relationship between DNA cross-linking, cell cycle perturbations, and apoptosis. J. Pharmacol. Exp. Ther. 2005, 313, 771–779. [Google Scholar] [CrossRef]
- Miliukienė, V.; Nivinskas, H.; Čėnas, N. Cytotoxicity of anticancer aziridinyl-substituted benzoquinones in primary mice splenocytes. Acta Biochim. Pol. 2014, 61, 833–836. [Google Scholar] [CrossRef]
- Ger, M.; Kaupinis, A.; Nemeikaite-Ceniene, A.; Sarlauskas, J.; Cicenas, J.; Cenas, N.; Valius, M. Quantitative proteomic analysis of anticancer drug RH1 resistance in liver carcinoma. Biochim. Biophys. Acta—Proteins Proteomics 2016, 1864, 219–232. [Google Scholar] [CrossRef]
- Winski, S.L.; Hargreaves, R.H.; Butler, J.; Ross, D. A new screening system for NAD(P)H:quinone oxidoreductase (NQO1)-directed antitumor quinones: Identification of a new aziridinylbenzoquinone, RH1, as a NQO1-directed antitumor agent. Clin. Cancer Res. 1998, 4, 3083–3088. [Google Scholar]
- Hasinoff, B.B.; Begleiter, A. The reductive activation of the antitumor drug RH1 to its semiquinone free radical by NADPH cytochrome P450 reductase and by HCT116 human colon cancer cells. Free Radic. Res. 2006, 40, 974–978. [Google Scholar] [CrossRef]
- Siegel, D.; Yan, C.; Ross, D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the sensitivity and resistance to antitumor quinones. Biochem. Pharmacol. 2012, 83, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Park, M.-T.; Choi, B.-H.; Oh, E.-T.; Song, M.-J.; Lee, J.; Kim, C.; Lim, B.U.; Park, H.J. Endoplasmic Reticulum Stress-Induced JNK Activation Is a Critical Event Leading to Mitochondria-Mediated Cell Death Caused by β-Lapachone Treatment. PLoS ONE 2011, 6, e21533. [Google Scholar] [CrossRef]
- Wu, K.; Knox, R.; Sun, X.Z.; Joseph, P.; Jaiswal, A.K.; Zhang, D.; Deng, P.S.-K.; Chen, S. Catalytic Properties of NAD(P)H:Quinone Oxidoreductase-2 (NQO2), a Dihydronicotinamide Riboside Dependent Oxidoreductase. Arch. Biochem. Biophys. 1997, 347, 221–228. [Google Scholar] [CrossRef]
- Stulpinas, A.; Imbrasaite, A.; Krestnikova, N.; Sarlauskas, J.; Cenas, N.; Kalvelyte, A.V. Study of Bioreductive Anticancer Agent RH-1-Induced Signals Leading the Wild-Type p53-Bearing Lung Cancer A549 Cells to Apoptosis. Chem. Res. Toxicol. 2016, 29, 26–39. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Montojo, J.; Zuberi, K.; Rodriguez, H.; Kazi, F.; Wright, G.; Donaldson, S.L.; Morris, Q.; Bader, G.D. GeneMANIA Cytoscape plugin: Fast gene function predictions on the desktop. Bioinformatics 2010, 26, 2927–2928. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2014, 11, 9–15. [Google Scholar] [CrossRef] [Green Version]
- King, T.D.; Suto, M.J.; Li, Y. The wnt/β-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. J. Cell. Biochem. 2012, 113, 13–18. [Google Scholar] [CrossRef]
- Holland, J.D.; Klaus, A.; Garratt, A.N.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Curr. Opin. Cell Biol. 2013, 25, 254–264. [Google Scholar] [CrossRef]
- Lenos, K.J.; Vermeulen, L. Cancer stem cells don’t waste their time cleaning-low proteasome activity, a marker for cancer stem cell function. Ann. Transl. Med. 2016, 4, 519. [Google Scholar] [CrossRef]
- Cooper, M.J.; Cox, N.J.; Zimmerman, E.I.; Dewar, B.J.; Duncan, J.S.; Whittle, M.C.; Nguyen, T.A.; Jones, L.S.; Ghose Roy, S.; Smalley, D.M.; et al. Application of Multiplexed Kinase Inhibitor Beads to Study Kinome Adaptations in Drug-Resistant Leukemia. PLoS ONE 2013, 8, e66755. [Google Scholar] [CrossRef]
- Wissing, J.; Jänsch, L.; Nimtz, M.; Dieterich, G.; Hornberger, R.; Kéri, G.; Wehland, J.; Daub, H. Proteomics Analysis of Protein Kinases by Target Class-selective Prefractionation and Tandem Mass Spectrometry. Mol. Cell. Proteomics 2007, 6, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Bantscheff, M.; Hopf, C.; Kruse, U.; Drewes, G. Proteomics-based strategies in kinase drug discovery. Ernst Schering Found. Symp. Proc. 2007, 3, 1–28. [Google Scholar]
- Duncan, J.; Whittle, M.; Nakamura, K.; Abell, A.; Midland, A.; Zawistowski, J.; Johnson, N.; Granger, D.; Jordan, N.; Darr, D.; et al. Dynamic Reprogramming of the Kinome in Response to Targeted MEK Inhibition in Triple-Negative Breast Cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Patricelli, M.; Nomanbhoy, T.; Wu, J.; Brown, H.; Zhou, D.; Zhang, J.; Jagannathan, S.; Aban, A.; Okerberg, E.; Herring, C.; et al. In Situ Kinase Profiling Reveals Functionally Relevant Properties of Native Kinases. Chem. Biol. 2011, 18, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Breitkopf, S.B.; Oppermann, F.S.; Ke?ri, G.; Grammel, M.; Daub, H. Proteomics Analysis of Cellular Imatinib Targets and their Candidate Downstream Effectors. J. Proteome Res. 2010, 9, 6033–6043. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Oppermann, F.S.; Gnad, F.; Olsen, J.V.; Hornberger, R.; Greff, Z.; Kéri, G.; Mann, M.; Daub, H. Large-scale Proteomics Analysis of the Human Kinome. Mol. Cell. Proteomics 2009, 8, 1751–1764. [Google Scholar] [CrossRef] [Green Version]
- Unwin, R.D.; Griffiths, J.R.; Whetton, A.D. Simultaneous analysis of relative protein expression levels across multiple samples using iTRAQ isobaric tags with 2D nano LC?MS/MS. Nat. Protoc. 2010, 5, 1574–1582. [Google Scholar] [CrossRef]
- Chang, T.-Y.; Chen, H.-A.; Chiu, C.-F.; Chang, Y.-W.; Kuo, T.-C.; Tseng, P.-C.; Wang, W.; Hung, M.-C.; Su, J.-L. Dicer Elicits Paclitaxel Chemosensitization and Suppresses Cancer Stemness in Breast Cancer by Repressing AXL. Cancer Res. 2016, 76, 3916–3928. [Google Scholar] [CrossRef] [Green Version]
- Corominas-Faja, B.; CUF?, S.; Oliveras-Ferraros, C.; CUY?S, E.; L?pez-Bonet, E.; LUPU, R.; ALARC?N, T.; Vellon, L.; Manuel Iglesias, J.; LEIS, O.; et al. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle 2013, 12, 3109–3124. [Google Scholar] [CrossRef] [Green Version]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Halabi, N.; Guerrouahen, B.S.; Rafii, S.; Rafii, A. Breast cancer cells promote a notch-dependent mesenchymal phenotype in endothelial cells participating to a pro-tumoral niche. J. Transl. Med. 2015, 13, 27. [Google Scholar] [CrossRef]
- Hebbard, L.W.; Maurer, J.; Miller, A.; Lesperance, J.; Hassell, J.; Oshima, R.G.; Terskikh, A.V. Maternal Embryonic Leucine Zipper Kinase Is Upregulated and Required in Mammary Tumor-Initiating Cells In vivo. Cancer Res. 2010, 70, 8863–8873. [Google Scholar] [CrossRef]
- Jeon, H.-M.; Lee, J. MET: Roles in epithelial-mesenchymal transition and cancer stemness. Ann. Transl. Med. 2017, 5, 5. [Google Scholar] [CrossRef]
- Pandolfi, S.; Stecca, B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: Implications for cancer therapy. Expert Rev. Mol. Med. 2015, 17, e5. [Google Scholar] [CrossRef]
- Parris, A.B.; Zhao, Q.; Howard, E.W.; Zhao, M.; Ma, Z.; Yang, X. Buformin inhibits the stemness of erbB-2-overexpressing breast cancer cells and premalignant mammary tissues of MMTV-erbB-2 transgenic mice. J. Exp. Clin. Cancer Res. 2017, 36, 28. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.-W.; Cheng, C.-C.; Yang, S.-T.; Tseng, C.-F.; Chang, T.-Y.; Tsai, S.-Y.; Fu, E.; Chiang, C.-P.; Liao, L.-C.; Tsai, P.-W.; et al. Targeting the VEGF-C/VEGFR3 axis suppresses Slug-mediated cancer metastasis and stemness via inhibition of KRAS/YAP1 signaling. Oncotarget 2017, 8, 5603–5618. [Google Scholar] [CrossRef]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E.; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [Green Version]
- Stulpinas, A.; Imbrasaitė, A.; Kalvelytė, A.V. Daunorubicin induces cell death via activation of apoptotic signalling pathway and inactivation of survival pathway in muscle-derived stem cells. Cell Biol. Toxicol. 2012, 28, 103–114. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef]
- Haikala, H.M.; Klefstrom, J.; Eilers, M.; Wiese, K.E. MYC-induced apoptosis in mammary epithelial cells is associated with repression of lineage-specific gene signatures. Cell Cycle 2016, 15, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Ferraris, S.E.; Isoniemi, K.; Torvaldson, E.; Anckar, J.; Westermarck, J.; Eriksson, J.E. Nucleolar AATF regulates c-Jun-mediated apoptosis. Mol. Biol. Cell 2012, 23, 4323–4332. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Park, J.-R.; Kim, E.-J.; Kim, W.J.; Hong, S.-H.; Park, S.-M.; Yang, S.-R. Cigarette smoke-mediated oxidative stress induces apoptosis via the MAPKs/STAT1 pathway in mouse lung fibroblasts. Toxicol. Lett. 2016, 240, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Hou, J.; Huang, C.; Sun, X.; Zeng, Y.; Cheng, H.; Wang, H.; Li, C. C-Myc regulates radiation-induced G2/M cell cycle arrest and cell death in human cervical cancer cells. J. Obstet. Gynaecol. Res. 2017, 43, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Chan, Q.K.Y.; Lam, H.-M.; Ng, C.-F.; Lee, A.Y.Y.; Chan, E.S.Y.; Ng, H.-K.; Ho, S.-M.; Lau, K.-M. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G2 cell-cycle arrest. Cell Death Differ. 2010, 17, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Dimco, G.; Knight, R.A.; Latchman, D.S.; Stephanou, A. STAT1 interacts directly with cyclin D1/Cdk4 and mediates cell cycle arrest. Cell Cycle 2010, 9, 4638–4649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.J.; Polack, A.; Dalla-Favera, R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 1999, 283, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.W.; Fang, M.; Park, S.M.; Hutchinson, L.; Green, M.R. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. Elife 2014, 3, e02313. [Google Scholar] [CrossRef] [PubMed]
- Reardon, C.; McKay, D.M. TGF-beta suppresses IFN-gamma-STAT1-dependent gene transcription by enhancing STAT1-PIAS1 interactions in epithelia but not monocytes/macrophages. J. Immunol. 2007, 178, 4284–4295. [Google Scholar] [CrossRef]
- Holaska, J.M.; Rais-Bahrami, S.; Wilson, K.L. Lmo7 is an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum. Mol. Genet. 2006, 15, 3459–3472. [Google Scholar] [CrossRef]
- Lykke-Andersen, J. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol. Cell. Biol. 2002, 22, 8114–8121. [Google Scholar] [CrossRef]
- Howell, B.; Larsson, N.; Gullberg, M.; Cassimeris, L. Dissociation of the tubulin-sequestering and microtubule catastrophe-promoting activities of oncoprotein 18/stathmin. Mol. Biol. Cell 1999, 10, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Li, F.; Ballif, B.A.; Li, S.; Chen, X.; Guo, L.; Ye, X. Identification and Functional Analysis of a Novel Cyclin E/Cdk2 Substrate Ankrd17. J. Biol. Chem. 2009, 284, 7875–7888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotipatruni, R.R.; Nalla, A.K.; Asuthkar, S.; Gondi, C.S.; Dinh, D.H.; Rao, J.S. Apoptosis Induced by Knockdown of uPAR and MMP-9 is Mediated by Inactivation of EGFR/STAT3 Signaling in Medulloblastoma. PLoS ONE 2012, 7, e44798. [Google Scholar] [CrossRef] [PubMed]
- Saxena, N.K.; Vertino, P.M.; Anania, F.A.; Sharma, D. Leptin-induced Growth Stimulation of Breast Cancer Cells Involves Recruitment of Histone Acetyltransferases and Mediator Complex to CYCLIN D1 Promoter via Activation of Stat3. J. Biol. Chem. 2007, 282, 13316–13325. [Google Scholar] [CrossRef] [PubMed]
- Numata, A.; Shimoda, K.; Kamezaki, K.; Haro, T.; Kakumitsu, H.; Shide, K.; Kato, K.; Miyamoto, T.; Yamashita, Y.; Oshima, Y.; et al. Signal Transducers and Activators of Transcription 3 Augments the Transcriptional Activity of CCAAT/Enhancer-binding Protein α in Granulocyte Colony-stimulating Factor Signaling Pathway. J. Biol. Chem. 2005, 280, 12621–12629. [Google Scholar] [CrossRef] [PubMed]
- Hajimoradi, M.; Mohammad Hassan, Z.; Ebrahimi, M.; Soleimani, M.; Bakhshi, M.; Firouzi, J.; Samani, F.S. STAT3 is Overactivated in Gastric Cancer Stem-Like Cells. Cell J. 2016, 17, 617–628. [Google Scholar] [PubMed]
- Tcherkezian, J.; Cargnello, M.; Romeo, Y.; Huttlin, E.L.; Lavoie, G.; Gygi, S.P.; Roux, P.P. Proteomic analysis of cap-dependent translation identifies LARP1 as a key regulator of 5′TOP mRNA translation. Genes Dev. 2014, 28, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.D.; Zakaria, C.; Jia, J.-J.; Graber, T.E.; Svitkin, Y.; Tahmasebi, S.; Healy, D.; Hoang, H.-D.; Jensen, J.M.; Diao, I.T.; et al. La-related Protein 1 (LARP1) Represses Terminal Oligopyrimidine (TOP) mRNA Translation Downstream of mTOR Complex 1 (mTORC1). J. Biol. Chem. 2015, 290, 15996–16020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, N.C.; Jefferies, H.B.J.; Alemu, E.A.; Saunders, R.E.; Howell, M.; Johansen, T.; Tooze, S.A. Genome-wide siRNA screen reveals amino acid starvation-induced autophagy requires SCOC and WAC. EMBO J. 2012, 31, 1931–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, T.G.; Mura, M.; Al-Ashtal, H.A.; Lahr, R.M.; Abd-Latip, N.; Sweeney, K.; Lu, H.; Weir, J.; El-Bahrawy, M.; Steel, J.H.; et al. The RNA-binding protein LARP1 is a post-transcriptional regulator of survival and tumorigenesis in ovarian cancer. Nucleic Acids Res. 2016, 44, 1227–1246. [Google Scholar] [CrossRef] [PubMed]
- Ozgur, S.; Chekulaeva, M.; Stoecklin, G. Human Pat1b Connects Deadenylation with mRNA Decapping and Controls the Assembly of Processing Bodies. Mol. Cell. Biol. 2010, 30, 4308–4323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totaro, A.; Renzi, F.; La Fata, G.; Mattioli, C.; Raabe, M.; Urlaub, H.; Achsel, T. The human Pat1b protein: A novel mRNA deadenylation factor identified by a new immunoprecipitation technique. Nucleic Acids Res. 2011, 39, 635–647. [Google Scholar] [CrossRef] [PubMed]
- De Morree, A.; Droog, M.; Grand Moursel, L.; Bisschop, I.J.M.; Impagliazzo, A.; Frants, R.R.; Klooster, R.; van der Maarel, S.M. Self-regulated alternative splicing at the AHNAK locus. FASEB J. 2012, 26, 93–103. [Google Scholar] [CrossRef]
- Bačević, K.; Lossaint, G.; Achour, T.N.; Georget, V.; Fisher, D.; Dulić, V. Cdk2 strengthens the intra-S checkpoint and counteracts cell cycle exit induced by DNA damage. Sci. Rep. 2017, 7, 13429. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef] [PubMed]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef]
- Huang, Y.; Zhou, L.; Meng, X.; Yu, B.; Wang, H.; Yang, Y.; Wu, Y.; Tan, X. IRS-1 Regulates Proliferation, Invasion and Metastasis of Pancreatic Cancer Cells through MAPK and PI3K Signaling Pathways. Int. J. Clin. Exp. Pathol. 2018, 11, 5185–5193. [Google Scholar]
- Concannon, C.G.; Gorman, A.M.; Samali, A. On the role of Hsp27 in regulating apoptosis. Apoptosis 2003, 8, 61–70. [Google Scholar] [CrossRef]
- Velier, J.; Kim, M.; Schwarz, C.; Kim, T.W.; Sapp, E.; Chase, K.; Aronin, N.; DiFiglia, M. Wild-Type and Mutant Huntingtins Function in Vesicle Trafficking in the Secretory and Endocytic Pathways. Exp. Neurol. 1998, 152, 34–40. [Google Scholar] [CrossRef]
- Feng, Y.; Walsh, C.A. The many faces of filamin: A versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 2004, 6, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Pierrat, B.; da Silva Correia, J.; Mary, J.-L.; Tomás-Zuber, M.; Lesslauer, W. RSK-B, a Novel Ribosomal S6 Kinase Family Member, Is a CREB Kinase under Dominant Control of p38α Mitogen-activated Protein Kinase (p38αMAPK). J. Biol. Chem. 1998, 273, 29661–29671. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.P.; Bastikar, V.A.; Kuciauskas, D.; Kothari, S.L.; Cicenas, J.; Valius, M. Molecular modeling and structure-based drug discovery approach reveals protein kinases as off-targets for novel anticancer drug RH1. Med. Oncol. 2017, 34, 176. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-T.; Song, M.-J.; Oh, E.-T.; Lee, H.; Choi, B.-H.; Jeong, S.-Y.; Choi, E.-K.; Park, H.J. The anti-tumour compound, RH1, causes mitochondria-mediated apoptosis by activating c-Jun N-terminal kinase. Br. J. Pharmacol. 2011, 163, 567–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, E.; Honeth, G.; Bendahl, P.-O.; Saal, L.H.; Gruvberger-Saal, S.; Ringnér, M.; Vallon-Christersson, J.; Jönsson, G.; Holm, K.; Lövgren, K.; et al. CD44 isoforms are heterogeneously expressed in breast cancer and correlate with tumor subtypes and cancer stem cell markers. BMC Cancer 2011, 11, 418. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Lorenzo, I.; Dorado, J.; Lonardo, E.; Alcala, S.; Serrano, A.G.; Clausell-Tormos, J.; Cioffi, M.; Megias, D.; Zagorac, S.; Balic, A.; et al. Intracellular autofluorescence: a biomarker for epithelial cancer stem cells. Nat. Methods 2014, 11, 1161–1169. [Google Scholar] [CrossRef]
- Foster, B.M.; Zaidi, D.; Young, T.R.; Mobley, M.E.; Kerr, B.A. CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance. Biomedicines 2018, 6, 31. [Google Scholar] [CrossRef]
- Tsang, J.Y.S.; Huang, Y.-H.; Luo, M.-H.; Ni, Y.-B.; Chan, S.-K.; Lui, P.C.W.; Yu, A.M.C.; Tan, P.H.; Tse, G.M. Cancer stem cell markers are associated with adverse biomarker profiles and molecular subtypes of breast cancer. Breast Cancer Res. Treat. 2012, 136, 407–417. [Google Scholar] [CrossRef]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and In vitro Propagation of Tumorigenic Breast Cancer Cells with Stem/Progenitor Cell Properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef]
- Trudeau, M.; Charbonneau, F.; Gelmon, K.; Laing, K.; Latreille, J.; Mackey, J.; McLeod, D.; Pritchard, K.; Provencher, L.; Verma, S. Selection of adjuvant chemotherapy for treatment of node-positive breast cancer. Lancet Oncol. 2005, 6, 886–898. [Google Scholar] [CrossRef]
- Roché, H.; Fumoleau, P.; Spielmann, M.; Canon, J.-L.; Delozier, T.; Serin, D.; Symann, M.; Kerbrat, P.; Soulié, P.; Eichler, F.; et al. Sequential Adjuvant Epirubicin-Based and Docetaxel Chemotherapy for Node-Positive Breast Cancer Patients: The FNCLCC PACS 01 Trial. J. Clin. Oncol. 2006, 24, 5664–5671. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, B.; Arnedos, M.; Popat, S.; Ashley, S.; Nerurkar, A.; Walsh, G.; Johnston, S.; Smith, I.E. Platinum-based chemotherapy in triple-negative breast cancer. Ann. Oncol. 2008, 19, 1847–1852. [Google Scholar] [CrossRef] [PubMed]
- Duncia, J.V.; Santella, J.B.; Higley, C.A.; Pitts, W.J.; Wityak, J.; Frietze, W.E.; Rankin, F.W.; Sun, J.H.; Earl, R.A.; Tabaka, A.C.; et al. MEK inhibitors: The chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg. Med. Chem. Lett. 1998, 8, 2839–2844. [Google Scholar] [CrossRef]
- Cicenas, J. The Aurora kinase inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. 2016, 142, 1995–2012. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J. STI571 (Gleevec) as a paradigm for cancer therapy. Trends Mol. Med. 2002, 8, S14–S18. [Google Scholar] [CrossRef]
- Levina, V.; Marrangoni, A.; Wang, T.; Parikh, S.; Su, Y.; Herberman, R.; Lokshin, A.; Gorelik, E. Elimination of Human Lung Cancer Stem Cells through Targeting of the Stem Cell Factor-c-kit Autocrine Signaling Loop. Cancer Res. 2010, 70, 338–346. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Varemo, L.; Nielsen, J.; Nookaew, I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Glatter, T.; Ludwig, C.; Ahrn?, E.; Aebersold, R.; Heck, A.J.R.; Schmidt, A. Large-Scale Quantitative Assessment of Different In-Solution Protein Digestion Protocols Reveals Superior Cleavage Efficiency of Tandem Lys-C/Trypsin Proteolysis over Trypsin Digestion. J. Proteome Res. 2012, 11, 5145–5156. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence | Reverse Sequence |
|---|---|---|
| DPP4 | 5′-AGTGGCACGGCAACACATT-3′ | 5′-AGAGCTTCTATCCCGATGACTT-3′ |
| CD44 | 5′-CTGCCGCTTTGCAGGTGTA-3′ | 5′-CATTGTGGGCAAGGTGCTATT-3′ |
| MSI | 5′-TAAAGTGCTGGCGCAATCG-3′ | 5′-TCTTCTTCGTTCGAGTCACCA-3′ |
| ALCAM | 5′-ACTTGACGTACCTCAGAATCTCA-3′ | 5′-CATCGTCGTACTGCACACTTT-3′ |
| PROM1 | 5′-TTCTTGACCGACTGAGACCCA-3′ | 5′-TCATGTTCTCCAACGCCTCTT-3′ |
| KIT | 5′-ACTTGAGGTTTATTCCTGACCCC-3′ | 5′-GCAGACAGAGCCGATGGTAG-3′ |
| KITLG | 5′-AATCCTCTCGTCAAAACTGAAGG-3′ | 5′-CCATCTCGCTTATCCAACAATGA-3′ |
| HPRT1 | 5′-TGCAGACTTTGCTTTCCTTGGTC-3′ | 5′-CCAACACTTCGTGGGGTCCTT-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuciauskas, D.; Dreize, N.; Ger, M.; Kaupinis, A.; Zemaitis, K.; Stankevicius, V.; Suziedelis, K.; Cicenas, J.; Graves, L.M.; Valius, M. Proteomic Analysis of Breast Cancer Resistance to the Anticancer Drug RH1 Reveals the Importance of Cancer Stem Cells. Cancers 2019, 11, 972. https://doi.org/10.3390/cancers11070972
Kuciauskas D, Dreize N, Ger M, Kaupinis A, Zemaitis K, Stankevicius V, Suziedelis K, Cicenas J, Graves LM, Valius M. Proteomic Analysis of Breast Cancer Resistance to the Anticancer Drug RH1 Reveals the Importance of Cancer Stem Cells. Cancers. 2019; 11(7):972. https://doi.org/10.3390/cancers11070972
Chicago/Turabian StyleKuciauskas, Dalius, Nadezda Dreize, Marija Ger, Algirdas Kaupinis, Kristijonas Zemaitis, Vaidotas Stankevicius, Kestutis Suziedelis, Jonas Cicenas, Lee M. Graves, and Mindaugas Valius. 2019. "Proteomic Analysis of Breast Cancer Resistance to the Anticancer Drug RH1 Reveals the Importance of Cancer Stem Cells" Cancers 11, no. 7: 972. https://doi.org/10.3390/cancers11070972