Resistance to Systemic Agents in Renal Cell Carcinoma Predict and Overcome Genomic Strategies Adopted by Tumor

 ,
,  ,
, 
 ,
,
Abstract
:1. Introduction
2. Resistance to Target Therapy
2.1. RCC Heterogeneity and Recurrent Mutations
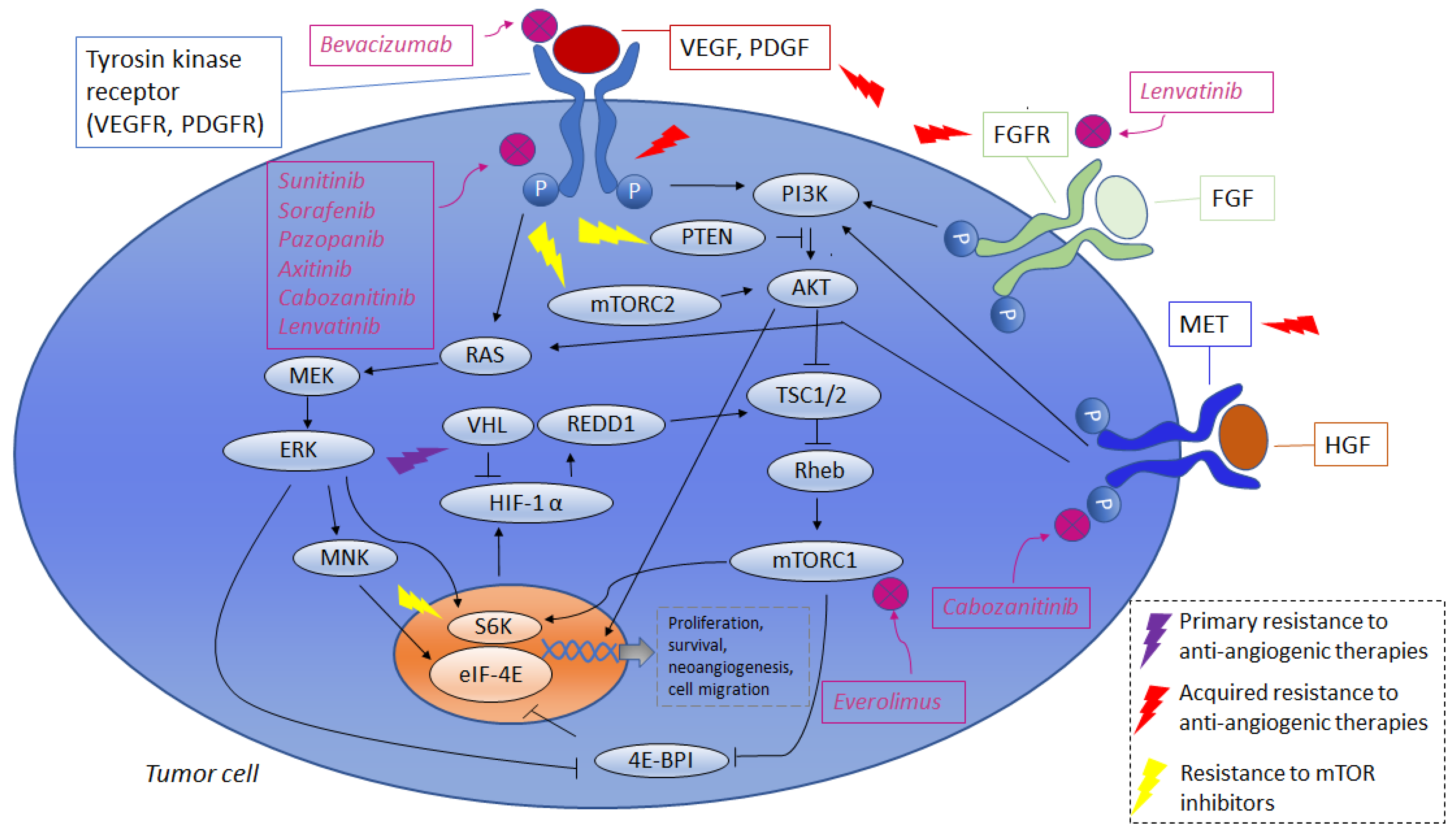
2.2. Primary Resistance to Agents Targeting Angiogenesis
2.3. Acquired Resistance to Agents Targeting Angiogenesis
3. Resistance to mTOR Inhibitors
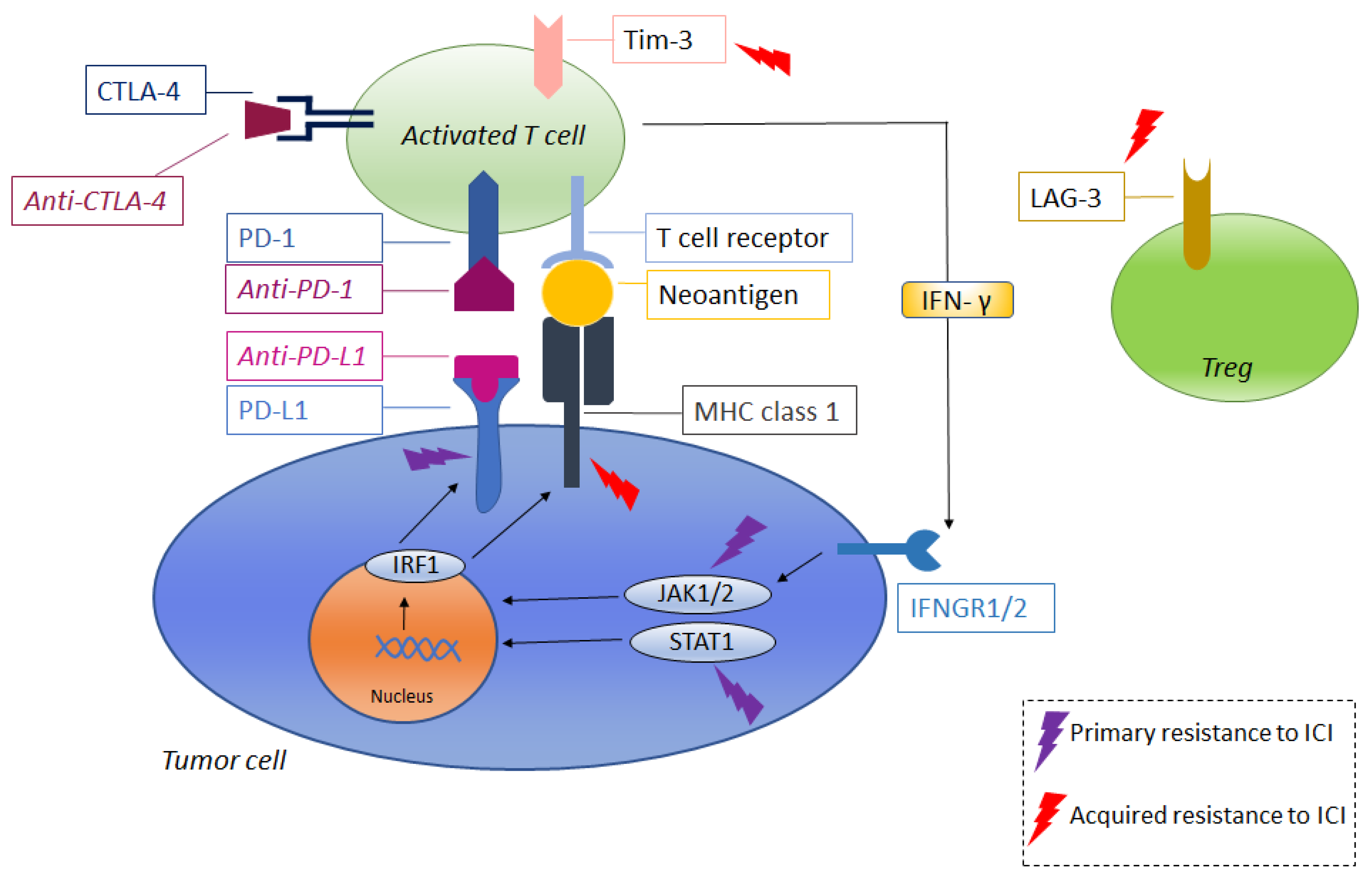
4. Resistance to Immune-Checkpoint Inhibitors
4.1. Primary Resistance to Immune-Checkpoint Inhibitors
4.2. Acquired Resistance to Immune-Checkpoint Inhibitors
5. Predicting Resistance and Sensitivity to Systemic Treatment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO classification of tumours of the urinary system and male genital organs—part A: Renal, penile, and testicular tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 3698. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.F.; Ricketts, C.J.; Wang, M.; Yang, L.; Cherniack, A.D.; Shen, H.; Buhay, C.; Kang, H.; Kim, S.C.; Fahey, C.C.; et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 2014, 26, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.H.; Choueiri, T.K.; Heng, D.Y.; Albiges, L.; Hsieh, J.J.; Linehan, W.M.; Pal, S.; Maskens, D.; Paseman, B.; Jonasch, E.; et al. Recommendations for the Management of Rare Kidney Cancers. Eur. Urol. 2017, 72, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Motzer, R.J. Systemic therapy for metastatic renal-cell carcinoma. N. Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef]
- Santoni, M.; Massari, F.; Piva, F.; Carrozza, F.; Di Nunno, V.; Cimadamore, A.; Martignetti, A.; Montironi, R.; Battelli, N. Tivozanib for the treatment of renal cell carcinoma. Expert Opin. Pharmacother. 2018, 19, 1021–1025. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Gore, M.E.; Szczylik, C.; Porta, C.; Bracarda, S.; Bjarnason, G.A.; Oudard, S.; Hariharan, S.; Lee, S.H.; Haanen, J.; Castellano, D.; et al. Safety and efficacy of sunitinib for metastatic renal-cell carcinoma: An expanded-access trial. Lancet Oncol. 2009, 10, 757–763. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, C.N.; Hawkins, R.E.; Wagstaff, J.; Salman, P.; Mardiak, J.; Barrios, C.H.; Zarba, J.J.; Gladkov, O.A.; Lee, E.; Szczylik, C.; et al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: Final overall survival results and safety update. Eur. J. Cancer 2013, 49, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R.; et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Tannir, N.M.; Mainwaring, P.N.; Rini, B.I.; Hammers, H.J.; Donskov, F.; Roth, B.J.; Peltola, K.; et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): Final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 917–927. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Tomczak, P.; Hutson, T.E.; Michaelson, M.D.; Negrier, S.; Oudard, S.; Gore, M.E.; Tarazi, J.; Hariharan, S.; et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: Overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013, 14, 552–562. [Google Scholar] [CrossRef]
- Massari, F.; Di Nunno, V.; Ciccarese, C.; Graham, J.; Porta, C.; Comito, F.; Cubelli, M.; Iacovelli, R.; Heng, D.Y.C. Adjuvant therapy in renal cell carcinoma. Cancer Treat. Rev. 2017, 60, 152–157. [Google Scholar] [CrossRef]
- Massari, F.; Di Nunno, V.; Mollica, V.; Graham, J.; Gatto, L.; Heng, D. Adjuvant Tyrosine Kinase Inhibitors in Treatment of Renal Cell Carcinoma: A Meta-Analysis of Available Clinical Trials. Clin. Genitourin Cancer 2019. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Ciccarese, C.; Di Nunno, V.; Iacovelli, R.; Massari, F. Future perspectives for personalized immunotherapy in renal cell carcinoma. Expert Opin. Biol. Ther. 2017, 17, 1049–1052. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Guerra, F.; Conti, A.; Lucarelli, A.; Rinaldi, S.; Belvederesi, L.; Capucci, A.; Berardi, R. Incidence and risk of cardiotoxicity in cancer patients treated with targeted therapies. Cancer Treat. Rev. 2017, 59, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lee, J.L.; Ahn, J.H.; Lee, K.H.; Jeong, I.G.; Song, C.; Hong, B.; Hong, J.H.; Ahn, H. Efficacy and safety of everolimus in korean patients with metastatic renal cell carcinoma following treatment failure with a vascular endothelial growth factor receptor-tyrosine kinase inhibitor. Cancer Res. Treat. 2014, 46, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Vidal, M.J.; Martínez Ortega, E.; Montesa Pino, A.; Pérez Valderrama, B.; Viciana, R. Management of adverse events of targeted therapies in normal and special patients with metastatic renal cell carcinoma. Cancer Metastasis Rev. 2012, 31 (Suppl. 1), S19–S27. [Google Scholar] [CrossRef] [PubMed]
- Inno, A.; Metro, G.; Bironzo, P.; Grimaldi, A.M.; Grego, E.; Di Nunno, V.; Picasso, V.; Massari, F.; Gori, S. Pathogenesis, clinical manifestations and management of immune checkpoint inhibitors toxicity. Tumori 2017, 103, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Manley, B.J.; Khan, N.; Gao, J.; Carlo, M.I.; Cheng, E.H. Overcome tumor heterogeneity-imposed therapeutic barriers through convergent genomic biomarker discovery: A braided cancer river model of kidney cancer. Semin. Cell Dev. Biol. 2016, 64, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef]
- Gerlinger Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Wei, E.Y.; Hsieh, J.J. A river model to map convergent cancer evolution and guide therapy in RCC. Nat. Rev. Urol. 2015, 12, 706–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Lal, P.; Dondeti, V.R.; Letrero, R.; Parekh, K.N.; Oquendo, C.E.; Greenberg, R.A.; Flaherty, K.T.; Rathmell, W.K.; Keith, B.; et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 2008, 14, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Sarkadi, B.; Homolya, L.; Szakacs, G.; Varadi, A. Human multidrug resistance ABCB and ABCG transporters: Participation in a chemoimmunity defense system. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef] [PubMed]
- Gobe, G.; Rubin, M.; Williams, G.; Sawczuk, I.; Buttyan, R. Apoptosis and expression of Bcl-2, Bcl-XL, and Bax in renal cell carcinomas. Cancer Investig. 2002, 20, 324–332. [Google Scholar] [CrossRef]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Gill, A.J.; Hes, O.; Papathomas, T.; Šedivcová, M.; Tan, P.H.; Agaimy, A.; Andresen, P.A.; Kedziora, A.; Clarkson, A.; Toon, C.W.; et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: A morphologically distinct entity: A clinicopathologic series of 36 tumors from 27 patients. Am. J. Surg. Pathol. 2014, 38, 1588–1602. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Sulpice, E.; Ding, S.; Muscatelli-Groux, B.; Bergé, M.; Han, Z.C.; Plouet, J.; Tobelem, G.; Merkulova-Rainon, T. Cross-talk between the VEGF-A and HGF signalling pathways in endothelial cells. Biol. Cell 2009, 101, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Di Nunno, V.; Cubelli, M.; Massari, F. The role of the MET/AXL pathway as a new target for multikinase inhibitors in renal cell carcinoma. Expert Rev. Precis. Med. Drug Dev. 2017, 3, 169–175. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; Hammers, H.; Hutson, T.E.; Lee, J.L.; Peltola, K.; et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Halabi, S.; Sanford, B.L.; Hahn, O.; Michaelson, M.D.; Walsh, M.K.; Feldman, D.R.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib Versus Sunitinib as Initial Targeted Therapy for Patients with Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. J. Clin. Oncol. 2017, 35, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Plimack, E.; Arkenau, H.T.; Jonasch, E.; Heng, D.Y.C.; Powles, T.; Frigault, M.M.; Clark, E.A.; Handzel, A.A.; Gardner, H.; et al. Biomarker-Based Phase II Trial of Savolitinib in Patients With Advanced Papillary Renal Cell Cancer. J. Clin. Oncol. 2017, 35, 2993–3001. [Google Scholar] [CrossRef] [PubMed]
- Tsimafeyeu, I.; Demidov, L.; Stepanova, E.; Wynn, N.; Ta, H. Overexpression of fibroblast growth factor receptors FGFR1 and FGFR2 in renal cell carcinoma. Scand J. Urol. Nephrol. 2011, 45, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.A.; Leong, H.S.; Pavia-Jimenez, A.; Fedyshyn, S.; Yang, J.; Kucejova, B.; Sivanand, S.; Spence, P.; Xie, X.J.; Peña-Llopis, S.; et al. Fibroblast Growth Factor Receptor-Dependent and Independent Paracrine Signaling by Sunitinib-Resistant Renal Cell Carcinoma. Mol. Cell. Biol. 2016, 36, 1836–1855. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Porta, C.; Vogelzang, N.J.; Sternberg, C.N.; Szczylik, C.; Zolnierek, J.; Kollmannsberger, C.; Rha, S.Y.; Bjarnason, G.A.; Melichar, B.; et al. Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 286–296. [Google Scholar] [CrossRef]
- Schmidinger, M. Third-line dovitinib in metastatic renal cell carcinoma. Lancet Oncol. 2014, 15, 245–246. [Google Scholar] [CrossRef]
- Jonker, D.J.; Rosen, L.S.; Sawyer, M.B.; de Braud, F.; Wilding, G.; Sweeney, C.J.; Jayson, G.C.; McArthur, G.A.; Rustin, G.; Goss, G.; et al. A phase I study to determine the safety, pharmacokinetics and pharmacodynamics of a dual VEGFR and FGFR inhibitor, brivanib, in patients with advanced or metastatic solid tumors. Ann. Oncol. 2011, 22, 1413–1419. [Google Scholar] [CrossRef]
- Eisen, T.; Joensuu, H.; Nathan, P.D.; Harper, P.G.; Wojtukiewicz, M.Z.; Nicholson, S.; Bahl, A.; Tomczak, P.; Pyrhonen, S.; Fife, K.; et al. Regorafenib for patients with previously untreated metastatic or unresectable renal-cell carcinoma: A single-group phase 2 trial. Lancet Oncol. 2012, 13, 1055–1062. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef]
- Ishibashi, K.; Haber, T.; Breuksch, I.; Gebhard, S.; Sugino, T.; Kubo, H.; Hata, J.; Koguchi, T.; Yabe, M.; Kataoka, M.; et al. Overriding TKI resistance of renal cell carcinoma by combination therapy with IL-6 receptor blockade. Oncotarget 2017, 8, 55230–55245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.; Galisteo, R.; Gutkind, J.S. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine acti- vation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J. Biol. Chem. 2009, 284, 6038–6042. [Google Scholar] [CrossRef] [PubMed]
- Poletto, V.; Rosti, V.; Biggiogera, M.; Guerra, G.; Moccia, F.; Porta, C. The role of endothelial colony forming cells in kidney cancer’s pathogenesis, and in resistance to anti-VEGFR agents and mTOR inhibitors: A speculative review. Crit. Rev. Oncol./Hematol. 2018, 132, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saijo, Y.; Tanaka, M.; Miki, M.; Usui, K.; Suzuki, T.; Maemondo, M.; Hong, X.; Tazawa, R.; Kikuchi, T.; Matsushima, K.; et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: In vivo analysis of tumor-stromal interaction. J. Immunol. 2002, 169, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Dennis, P.B.; Jaeschke, A.; Saitoh, M.; Fowler, B.; Kozma, S.C.; Thomas, G. Mammalian TOR: A homeostatic ATP sensor. Science 2001, 294, 1102–1105. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. MTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Yoon, S.O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villén, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin–PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef]
- Figlin, R.A.; de Souza, P.; McDermott, D.; Dutcher, J.P.; Berkenblit, A.; Thiele, A.; Krygowski, M.; Strahs, A.; Feingold, J.; Boni, J.; et al. Analysis of PTEN and HIF-1alpha and correlation with efficacy in patients with advanced renal cell carcinoma treat- ed with temsirolimus versus interferon-alpha. Cancer 2009, 115, 3651–3660. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Pantano, F.; Amantini, C.; Nabissi, M.; Conti, A.; Burattini, L.; Zoccoli, A.; Berardi, R.; Santoni, G.; Tonini, G.; et al. Emerging strategies to overcome the resistance to current mTOR inhibitors in renal cell carcinoma. Biochim. Biophys. Acta 2014, 1845, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Okoh, V.O.; Felty, Q.; Parkash, J.; Poppiti, R.; Roy, D. Reactive oxygen species via redox signaling to PI3K/AKT pathway contribute to the malignant growth of 4-hydroxy estradiol-transformed mammary epithelial cells. PLoS ONE 2013, 8, e54206. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-g pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic b-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736.e18–749.e18. [Google Scholar] [CrossRef] [PubMed]
- Bernal, M.; Ruiz-Cabello, F.; Concha, A.; Paschen, A.; Garrido, F. Implication of the β2-microglobulin gene in the generation of tumor escape phenotypes. Cancer Immunol. Immunother. 2012, 61, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3-potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Knox, J.J.; Barrios, C.H.; Kim, T.M.; Cosgriff, T.; Srimuninnimit, V.; Pittman, K.; Sabbatini, R.; Rha, S.Y.; Flaig, T.W.; Page, R.D.; et al. Final overall survival analysis for the phase II RECORD-3 study of first-line everolimus followed by sunitinib versus first-line sunitinib followed by everolimus in metastatic RCC. Ann. Oncol. 2017, 28, 1339–1345. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targe ng Tim- 3 and PD- 1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef]
- Molina, A.M.; Hutson, T.E.; Nosov, D.; Tomczak, P.; Lipatov, O.; Sternberg, C.N.; Motzer, R.; Eisen, T. Efficacy of tivozanib treatment after sorafenib in patients with advanced renal cell carcinoma: Crossover of a phase 3 study. Eur. J. Cancer 2018, 94, 87–94. [Google Scholar] [CrossRef]
- Escudier, B.; Porta, C.; Bono, P.; Powles, T.; Eisen, T.; Sternberg, C.N.; Gschwend, J.E.; De Giorgi, U.; Parikh, O.; Hawkins, R.; et al. Randomized, controlled, double-blind, cross-over trial assessing treatment preference for pazopanib versus sunitinib in patients with metastatic renal cell carcinoma: PISCES Study. J. Clin. Oncol. 2014, 32, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Heng, D.Y.; Xie, W.; Regan, M.M.; Warren, M.A.; Golshayan, A.R.; Sahi, C.; Eigl, B.J.; Ruether, J.D.; Cheng, T.; North, S.; et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: Results from a large, multicenter study. J. Clin. Oncol. 2009, 27, 5794–5799. [Google Scholar] [CrossRef] [PubMed]
- Massari, F.; Di Nunno, V. CheckMate 214 patient-reported outcomes: Listening to our patients. Lancet Oncol. 2019, 20, 179–180. [Google Scholar] [CrossRef]
- Di Nunno, V.; Gatto, L.; Fragomeno, B.; Cubelli, M.; Nobili, E.; Romano, I.; Santoni, M.; Pisconti, S.; Montironi, R.; Massari, F. Combination immunotherapy in metastatic renal cell carcinoma. Are we leaving something back? Future Oncol. 2018, 14, 2997–2999. [Google Scholar] [CrossRef] [PubMed]
- Mollica, V.; Di Nunno, V.; Gatto, L.; Santoni, M.; Cimadamore, A.; Cheng, L.; Lopez-Beltran, A.; Montironi, R.; Pisconti SBattelli, N.; Massari, F. Novel Therapeutic Approaches and Targets Currently Under Evaluation for Renal Cell Carcinoma: Waiting for the Revolution. Clin. Drug Investig. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Nunno, V.D.; Mollica, V.; Gatto, L.; Santoni, M.; Cosmai, L.; Porta, C.; Massari, F. Prognostic impact of neutrophil-to-lymphocyte ratio in renal cell carcinoma: A systematic review and meta-analysis. Immunotherapy 2019, 11, 631–643. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Primary Resistance to Agents Targeting Angiogenesis | |
| Missed expression of targets | Tumors with wild type VHL alleles without HIF-α [33] |
| Cellular intake of target agents | Alteration in cell-surface proteins responsible of drugs intake [34] |
| Apoptosis inhibition | Increased expression of (Bcl-2/XL) [35] |
| Acquired Resistance to Agents Targeting Angiogenesis | |
| Acquisition of novel pathways promoting angiogenesis | - PDGFR [36] - MET [37,38,39,40] - FGFR [44,45] |
| Interactions with immune-system | - IL-8 promotes VEGF mRNA transcription and VEGFR-2 activation [51,52] - IL-6 promotes AKT/mTOR and STAT3 cascade promoting VEGF expression [51,52] |
| Angiogenesis induction by interleukin | - IL-1α and IL-1β induce angiogenesis. IL-1β may stimulate production of HIF-1α and VEGF [54,55] |
| Primary Resistance to mTOR Inhibitors | |
| Reactive oxygen species | - Increased levels of ROS may activate AKT pathways [62,63] |
| Acquired Resistance to mTOR Inhibitors | |
| Increased AKT activation mediated by mTORC1 inhibition. | - Inhibition of mTORC1 leads to reduced mTORC2 phosphorylation. Increased activity of mTORC 2 resulting from reduced phosphorylation leads to AKT activation [49,50] - mTORC1 inhibition results in missed GRB10 and S6K1 activation. These proteins exert negative feedback on AKT activation [59,60] - ERK/MAPK activation may be promoted by mTORC1 inhibition and leads to PI3K/AKT activation [61,62] |
| Primary Resistance to Immune-Checkpoint Inhibitors | |
| Reduced response to INFγ | - Continuous INFγ exposure may lead to downregulation or mutations on pathways related to INFγ response (for example Janus kinases JAK1/2, STATs) [66,67] |
| Reduced expression of INFγ and other genes related to immune-response | - PTEN loss may leads to INFγ or Granzyme B reduced expression [68] |
| Reduced T-Cells activity | - Activation of MAPK pathways leads to increased levels of VEGF and IL-8. This last interleukin has an inhibitory function on T-Cell activity [69] - Increased levels of β-catenin may lead to the loss of a specific subset of dendritic cells (CD103) resulting in resistance to immune-checkpoint inhibitors [70] |
| Reduced antigen production | - Mediated by sub-clones selection and epigenetic modification [65] |
| Balance between cells promoting and inhibiting immune-response | - Increased proportion of Tregs leads to the production of molecules inhibiting immune-response [71] - Immune-tumor contexture enriched of exhausted T effector cells and tumor-associated macrophages [72] |
| Acquired Resistance to Immune-Checkpoint Inhibitors | |
| Mechanisms involved in primary resistance | - Reduced response to INFγ - Reduced expression of INFγ and other genes related to immune-response - Reduced T-Cells activity - Reduced antigen production - Balance between cells promoting and inhibiting immune-response |
| Reduced MHC expression | - reduced expression of beta-2-microglobulin [73,75] |
| Interaction of other immune-checkpoints | - LAG3 [75,76] - TIM3 [77,79] |
| Study/First Author | Year | Experimental/Comparator Arm | ORR % PD as Best Response (Tumor with Primary Resistance) |
|---|---|---|---|
| First Line | |||
| NCT00098657 Motzer et al. [10] | 2007 | Sunitinib vs. Interferon | ORR = 31% PD = 21% |
| NCT00130897 Gore et al. [11] | 2009 | Sunitinib | ORR = 17% PD = 24% |
| COMPARZ Motzer et al. [14] | 2013 | Sunitinib vs. Pazopanib | ORR (S) = 25% PD (S) = 19% ORR (P) = 31% PD (P) = 17% |
| CHECKMATE 214 Motzer et al. [21] | 2018 | Nivolumab + Ipilimumab vs. Sunitinib | ORR (N + I) = 42% PD (N + I) = 20% ORR (S) = 27% PD (S) = 17% * |
| KEYNOTE 426 Rini et al. [22] | 2019 | Pembrolizumab + Axitinib vs. Sunitinib | ORR (P + A) = 59% PD (P + A) = 11% ORR (S) = 36% PD (S) = 17% |
| JAVELIN RENAL 101 Motzer et al. [23] | 2019 | Avelumab + Axitinib vs. Sunitinib | ORR (A + A) = 51% PD (A + A) = 11% ORR (S) = 26% PD (S) = 22% |
| Second Line | |||
| METEOR Choueiri et al. [41] | 2015 | Cabozantinib vs. Everolimus | ORR (C) = 21% PD (C) = 14% ORR (E) = 5% PD (E) = 27% |
| CHECKMATE 025 Motzer et al. [19] | 2015 | Nivolumab vs. Everolimus | ORR (N) = 25% PD (N) = 35% ORR (E) = 5% PD (E) = 28% |
| NCT00678392 Motzer et al. [16] | 2013 | Axitinib vs. Sorafenib | ORR (A) = 23 PD (A) = Not reported ORR (So) = 12 PD (S) = Not reported |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollica, V.; Di Nunno, V.; Gatto, L.; Santoni, M.; Scarpelli, M.; Cimadamore, A.; Lopez-Beltran, A.; Cheng, L.; Battelli, N.; Montironi, R.; et al. Resistance to Systemic Agents in Renal Cell Carcinoma Predict and Overcome Genomic Strategies Adopted by Tumor. Cancers 2019, 11, 830. https://doi.org/10.3390/cancers11060830
Mollica V, Di Nunno V, Gatto L, Santoni M, Scarpelli M, Cimadamore A, Lopez-Beltran A, Cheng L, Battelli N, Montironi R, et al. Resistance to Systemic Agents in Renal Cell Carcinoma Predict and Overcome Genomic Strategies Adopted by Tumor. Cancers. 2019; 11(6):830. https://doi.org/10.3390/cancers11060830
Chicago/Turabian StyleMollica, Veronica, Vincenzo Di Nunno, Lidia Gatto, Matteo Santoni, Marina Scarpelli, Alessia Cimadamore, Antonio Lopez-Beltran, Liang Cheng, Nicola Battelli, Rodolfo Montironi, and et al. 2019. "Resistance to Systemic Agents in Renal Cell Carcinoma Predict and Overcome Genomic Strategies Adopted by Tumor" Cancers 11, no. 6: 830. https://doi.org/10.3390/cancers11060830