Mechanisms behind Temsirolimus Resistance Causing Reactivated Growth and Invasive Behavior of Bladder Cancer Cells In Vitro

 ,
, 
Abstract
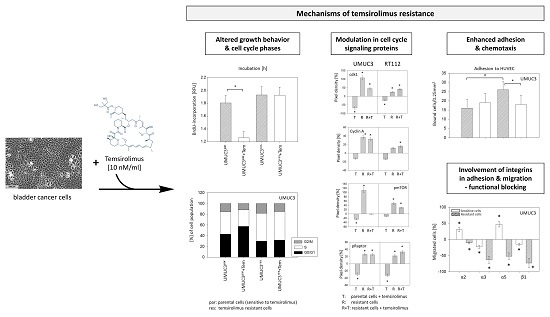
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Temsirolimus Resistance Alters Cell Growth, Proliferation, and Cell Cycling
2.2. Temsirolimus Resistance is Associated with Alterations of Cell Cycle Protein Expression
2.3. Temsirolimus Modifies Akt-mTOR-Signalling
2.4. Adhesion and Migration Properties of Resistant Versus Parental Cells
2.5. Temsirolimus Resistance is Accompanied by an Altered Integrin Expression Profile
2.6. Integrin Blocking Studies
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Temsirolimus Treatment
4.2. Measurement of Tumor Cell Growth, Proliferation, and Apoptosis
4.3. Analysis of Cell Cycling
4.4. Tumor Cell Adhesion to Vascular Endothelial Cells
4.5. Attachment to Immobilized Extracellular Matrix Proteins
4.6. Chemotactic Activity
4.7. Expression of Cell Cycle Regulating and Signalling Proteins
4.8. Integrin Surface and Protein Expression
4.9. Blocking Studies
4.10. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Retz, M.; Gschwend, J.E.; Maisch, P. Short version of the German S3 guideline for bladder cancer. Urologe A 2016, 55, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Massari, F.; Di Nunno, V.; Cubelli, M.; Santoni, M.; Fiorentino, M.; Montironi, R.; Cheng, L.; Lopez-Beltran, A.; Battelli, N.; Ardizzoni, A. Immune checkpoint inhibitors for metastatic bladder cancer. Cancer Treat. Rev. 2018, 64, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.Y.; Zeng, Q.H.; Cao, P.G.; Xie, D.; Yang, F.; He, L.Y.; Dai, Y.B.; Li, J.J.; Liu, X.M.; Zeng, H.L.; et al. SPAG5 promotes proliferation and suppresses apoptosis in bladder urothelial carcinoma by upregulating Wnt3 via activating the AKT/mTOR pathway and predicts poorer survival. Oncogene 2018, 37, 3937–3952. [Google Scholar] [CrossRef] [PubMed]
- Guancial, E.A.; Rosenberg, J.E. The role of genomics in the management of advanced bladder cancer. Curr. Treat. Options Oncol. 2015, 16, 319. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Greene, P.S.; Nielsen, M.E. Surgical advances in bladder cancer: At what cost? Urol. Clin. N. Am. 2015, 42, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Alfred Witjes, J.; Lebret, T.; Compérat, E.M.; Cowan, N.C.; De Santis, M.; Bruins, H.M.; Hernández, V.; Espinós, E.L.; Dunn, J.; Rouanne, M.; et al. Updated 2016 EAU Guidelines on Muscle-invasive and Metastatic Bladder Cancer. Eur. Urol. 2017, 71, 462–475. [Google Scholar] [CrossRef]
- von der Maase, H.; Sengelov, L.; Roberts, J.T.; Ricci, S.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Zimmermann, A.; Arning, M. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 2005, 23, 4602–4608. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; Théodore, C.; Demkov, T.; Komyakov, B.; Sengelov, L.; Daugaard, G.; Caty, A.; Carles, J.; Jagiello-Gruszfeld, A.; Karyakin, O.; et al. Phase III trial of vinflunine plus best supportive care compared with best supportive care alone after a platinum-containing regimen in patients with advanced transitional cell carcinoma of the urothelial tract. J. Clin. Oncol. 2009, 27, 4454–4461. [Google Scholar] [CrossRef]
- Spiess, P.E.; Agarwal, N.; Bangs, R.; Boorjian, S.A.; Buyyounouski, M.K.; Clark, P.E.; Downs, T.M.; Efstathiou, J.A.; Flaig, T.W.; Friedlander, T.; et al. Bladder Cancer, Version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 1240–1267. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.T.; Hui, G.; Mathis, C.; Chamie, K.; Pantuck, A.J.; Drakaki, A. The Current Status and Future Role of the Phosphoinositide 3 Kinase/AKT Signaling Pathway in Urothelial Cancer: An Old Pathway in the New Immunotherapy Era. Clin. Genitourin. Cancer 2018, 16, e269–e276. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network Nature. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, G..; Al-Ahmadie, H.; Schultz, N.; Hanrahan, A.J.; Ostrovnaya, I.; Balar, A.V.; Kim, P.H.; Lin, O.; Weinhold, N.; Sander, C.; et al. Prevalence and co-occurrence of actionable genomic alterations in high-grade bladder cancer. J. Clin. Oncol. 2013, 31, 3133–3140. [Google Scholar] [CrossRef] [PubMed]
- Winters, B.R.; Vakar-Lopez, F.; Brown, L.; Montgomery, B.; Seiler, R.; Black, P.C.; Boormans, J.L.; Dall Era, M.; Davincioni, E.; Douglas, J.; et al. Mechanistic target of rapamycin (MTOR) protein expression in the tumor and its microenvironment correlates with more aggressive pathology at cystectomy. Urol. Oncol. 2018, 36, 342. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Lee, T.J.; Chang, I.H. Role of the mTOR Pathway in the Progression and Recurrence of Bladder Cancer: An Immunohistochemical Tissue Microarray Study. Korean J. Urol. 2011, 52, 466–473. [Google Scholar] [CrossRef]
- Sun, C.H.; Chang, Y.H.; Pan, C.C. Activation of the PI3K/Akt/mTOR pathway correlates with tumour progression and reduced survival in patients with urothelial carcinoma of the urinary bladder. Histopathology 2011, 58, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Yuge, K.; Kikuchi, E.; Hagiwara, M.; Yasumizu, Y.; Tanaka, N.; Kosaka, T.; Miyajima, A.; Oya, M. Nicotine Induces Tumor Growth and Chemoresistance through Activation of the PI3K/Akt/mTOR Pathway in Bladder Cancer. Mol. Cancer Ther. 2015, 14, 2112–2120. [Google Scholar] [CrossRef]
- Pulido, M.; Roubaud, G.; Cazeau, A.L.; Mahammedi, H.; Vedrine, L.; Joly, F.; Mourey, L.; Pfister, C.; Goberna, A.; Lortal, B.; et al. Safety and efficacy of temsirolimus as second line treatment for patients with recurrent bladder cancer. BMC Cancer 2018, 18, 194. [Google Scholar] [CrossRef]
- Lau, D.K.; Tay, R.Y.; Yeung, Y.H.; Chionh, F.; Mooi, J.; Murone, C.; Skrinos, E.; Price, T.J.; Mariadason, J.M.; Tebbutt, N.C. Phase II study of everolimus (RAD001) monotherapy as first-line treatment in advanced biliary tract cancer with biomarker exploration: The RADiChol Study. Br. J. Cancer 2018, 118, 966–971. [Google Scholar] [CrossRef]
- Sathe, A.; Chalaud, G.; Oppolzer, I.; Wong, K.Y.; von Busch, M.; Schmid, S.C.; Tong, Z.; Retz, M.; Gschwend, J.E.; Schulz, W.A.; et al. Parallel PI3K, AKT and mTOR inhibition is required to control feedback loops that limit tumor therapy. PLoS ONE 2018, 13, e0190854. [Google Scholar] [CrossRef]
- Tsaur, I.; Makarević, J.; Hudak, L.; Juengel, E.; Kurosch, M.; Wiesner, C.; Bartsch, G.; Harder, S.; Haferkamp, A.; Blaheta, R.A. The cdk1-cyclin B complex is involved in everolimus triggered resistance in the PC3 prostate cancer cell line. Cancer Lett. 2011, 313, 84–90. [Google Scholar] [CrossRef]
- Juengel, E.; Nowaz, S.; Makarevi, J.; Natsheh, I.; Werner, I.; Nelson, K.; Reiter, M.; Tsaur, I.; Mani, J.; Harder, S.; et al. HDAC-inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol. Cancer 2014, 13, 152. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Hsu, I.; Sanford, T.; Railkar, R.; Balaji, N.; Sourbier, C.; Vocke, C.; Balaji, K.C.; Agarwal, P.K. Protein kinase D inhibitor CRT0066101 suppresses bladder cancer growth in vitro and xenografts via blockade of the cell cycle at G2/M. Cell. Mol. Life Sci. 2018, 75, 939–963. [Google Scholar] [CrossRef] [PubMed]
- Makarević, J.; Rutz, J.; Juengel, E.; Kaulfuss, S.; Reiter, M.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Amygdalin blocks bladder cancer cell growth in vitro by diminishing cyclin A and cdk2. PLoS ONE 2014, 9, e105590. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; You, S.; Oh, J.W.; Yoon, J.; Yeon, A.; Shahid, M.; Cho, E.; Sairam, V.; Park, T.D.; Kim, K.P.; et al. Integrated proteomic and phosphoproteomic analyses of cisplatin-sensitive and resistant bladder cancer cells reveal CDK2 network as a key therapeutic target. Cancer Lett. 2018, 437, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Liu, Y.B.; Zhou, Z.W.; Lian, B.; Li, H.; Li, J.P.; Zhou, S.F. miR-3646 promotes cell proliferation, migration, and invasion via regulating G2/M transition in human breast cancer cells. Am. J. Transl. Res. 2016, 8, 1659–1677. [Google Scholar] [PubMed]
- Garcia-España, A.; Salazar, E.; Sun, T.T.; Wu, X.R.; Pellicer, A. Differential expression of cell cycle regulators in phenotypic variants of transgenically induced bladder tumors: Implications for tumor behavior. Cancer Res. 2005, 65, 1150–1157. [Google Scholar] [CrossRef]
- Gao, Q.; Zheng, J. microRNA-323 upregulation promotes prostate cancer growth and docetaxel resistance by repressing p73. Biomed. Pharmacother. 2018, 97, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Kashiwagi, E.; Li, Y.; Zheng, Y.; Miyamoto, Y.; Netto, G.J.; Ishiguro, H.; Miyamoto, H. Cyclosporine A and tacrolimus inhibit urothelial tumorigenesis. Mol. Carcinog. 2016, 55, 161–169. [Google Scholar] [CrossRef]
- Blanca, A.; Requena, M.J.; Alvarez, J.; Cheng, L.; Montironi, R.; Raspollini, M.R.; Reymundo, C.; Lopez-Beltran, A. FGFR3 and Cyclin D3 as urine biomarkers of bladder cancer recurrence. Biomark. Med. 2016, 10, 243–253. [Google Scholar] [CrossRef]
- Shan, G.; Tang, T. Expression of cyclin D1 and cyclin E in urothelial bladder carcinoma detected in tissue chips using a quantum dot immunofluorescence technique. Oncol. Lett. 2015, 10, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yue, P.; Kim, Y.A.; Fu, H.; Khuri, F.R.; Sun, S.Y. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008, 68, 7409–7418. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.S. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Makarević, J.; Rutz, J.; Juengel, E.; Maxeiner, S.; Mani, J.; Vallo, S.; Tsaur, I.; Roos, F.; Chun, F.K.; Blaheta, R.A. HDAC Inhibition Counteracts Metastatic Re-Activation of Prostate Cancer Cells Induced by Chronic mTOR Suppression. Cells 2018, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhuang, J.; Deng, Y.; Zhao, X.; Tang, B.; Yao, D.; Zhao, W.; Chang, C.; Lu, Q.; Chen, W.; et al. GOLPH3 is a potential therapeutic target and a prognostic indicator of poor survival in bladder cancer treated by cystectomy. Oncotarget 2015, 6, 32177–32192. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Sharma, S. Mammalian target of rapamycin (mTOR) as a potential therapeutic target in various diseases. Inflammopharmacology 2017, 25, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Leite, R.; Arantes-Rodrigues, R.; Sousa, N.; Oliveira, P.A.; Santos, L. mTOR inhibitors in urinary bladder cancer. Tumour. Biol. 2016, 37, 11541–11551. [Google Scholar] [CrossRef] [PubMed]
- Vandekerkhove, G.; Todenhöfer, T.; Annala, M.; Struss, W.J.; Wong, A.; Beja, K.; Ritch, E.; Brahmbhatt, S.; Volik, S.V.; Hennenlotter, J.; et al. Circulating Tumor DNA Reveals Clinically Actionable Somatic Genome of Metastatic Bladder Cancer. Clin. Cancer Res. 2017, 23, 6487–6497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Cheng, S.; Zhang, Y.; Li, H.; Huang, J.; Zhang, P. Association between polymorphisms in the integrin gene predicted microRNA binding sites and bladder cancer risk. Int. J. Clin. Exp. Med. 2014, 7, 4398–4405. [Google Scholar]
- Engl, T.; Rutz, J.; Maxeiner, S.; Fanguen, S.; Juengel, E.; Koschade, S.; Roos, F.; Khoder, W.; Tsaur, I.; Blaheta, R.A. Acquired resistance to temsirolimus is associated with integrin α7 driven chemotactic activity of renal cell carcinoma in vitro. Oncotarget 2018, 9, 18747–18759. [Google Scholar] [CrossRef]
- Juengel, E.; Makarević, J.; Reiter, M.; Mani, J.; Tsaur, I.; Bartsch, G.; Haferkamp Blaheta, R.A. Resistance to the mTOR inhibitor temsirolimus alters adhesion and migration behavior of renal cell carcinoma cells through an integrin α5- and integrin β3-dependent mechanism. Neoplasia 2014, 16, 291–300. [Google Scholar] [CrossRef]
- Wedel, S.; Hudak, L.; Seibel, J.M.; Makarević, J.; Juengel, E.; Tsaur, I.; Wiesner, C.; Haferkamp, A.; Blaheta, R.A. Impact of combined HDAC and mTOR inhibition on adhesion, migration and invasion of prostate cancer cells. Clin. Exp. Metastasis 2011, 28, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Tsaur, I.; Makarević, J.; Juengel, E.; Gasser, M.; Waaga-Gasser, A.M.; Kurosch, M.; Reiter, M.; Wedel, S.; Bartsch, G.; Haferkamp, A.; et al. Resistance to the mTOR-inhibitor RAD001 elevates integrin α2- and β1-triggered motility, migration and invasion of prostate cancer cells. Br. J. Cancer 2012, 107, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wang, R.; Ren, G.; Li, X.; Wang, J.; Sun, Y.; Liang, J.; Nie, Y.; Wu, K.; Feng, B.; et al. HMGA2-FOXL2 Axis Regulates Metastases and Epithelial-to-Mesenchymal Transition of Chemoresistant Gastric Cancer. Clin. Cancer Res. 2017, 23, 3461–3473. [Google Scholar] [CrossRef]
- Sa, K.D.; Zhang, X.; Li, X.F.; Gu, Z.P.; Yang, A.G.; Zhang, R.; Li, J.P.; Sun, J.Y. A miR-124/ITGA3 axis contributes to colorectal cancer metastasis by regulating anoikis susceptibility. Biochem. Biophys. Res. Commun. 2018, 501, 758–764. [Google Scholar] [CrossRef]
- Xu, Z.; Zou, L.; Ma, G.; Wu, X.; Huang, F.; Feng, T.; Li, S.; Lin, Q.; He, X.; Liu, Z.; et al. Integrin β1 is a critical effector in promoting metastasis and chemo-resistance of esophageal squamous cell carcinoma. Am. J. Cancer Res. 2017, 7, 531–542. [Google Scholar] [PubMed]
- Carbonell, W.S.; DeLay, M.; Jahangiri, A.; Park, C.C.; Aghi, M.K. β1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013, 73, 3145–3154. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Qin, L.; Zhu, Z.; Wang, X.; Liu, Y.; Fan, Y.; Zhong, S.; Wang, X.; Zhang, X.; Xia, L.; et al. MicroRNA-31 functions as a tumor suppressor and increases sensitivity to mitomycin-C in urothelial bladder cancer by targeting integrin α5. Oncotarget 2016, 7, 27445–27457. [Google Scholar] [CrossRef]
- Fang, Z.; Yao, W.; Xiong, Y.; Zhang, J.; Liu, L.; Li, J.; Zhang, C.; Wan, J. Functional elucidation and methylation-mediated downregulation of ITGA5 gene in breast cancer cell line MDA-MB-468. J. Cell. Biochem. 2010, 110, 1130–1141. [Google Scholar] [CrossRef]
- Wang, H.; Shao, X.; He, Q.; Wang, C.; Xia, L.; Yue, D.; Qin, G.; Jia, C.; Chen, R. Quantitative Proteomics Implicates Rictor/mTORC2 in Cell Adhesion. J. Proteome Res. 2018, 17, 3360–3369. [Google Scholar] [CrossRef]
- Leng, C.; Zhang, Z.G.; Chen, W.X.; Luo, H.P.; Song, J.; Dong, W.; Zhu, X.R.; Chen, X.P.; Liang, H.F.; Zhang, B.X. An integrin beta4-EGFR unit promotes hepatocellular carcinoma lung metastases by enhancing anchorage independence through activation of FAK-AKT pathway. Cancer Lett. 2016, 376, 188–196. [Google Scholar] [CrossRef]














© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juengel, E.; Natsheh, I.; Najafi, R.; Rutz, J.; Tsaur, I.; Haferkamp, A.; Chun, F.K.-H.; Blaheta, R.A. Mechanisms behind Temsirolimus Resistance Causing Reactivated Growth and Invasive Behavior of Bladder Cancer Cells In Vitro. Cancers 2019, 11, 777. https://doi.org/10.3390/cancers11060777
Juengel E, Natsheh I, Najafi R, Rutz J, Tsaur I, Haferkamp A, Chun FK-H, Blaheta RA. Mechanisms behind Temsirolimus Resistance Causing Reactivated Growth and Invasive Behavior of Bladder Cancer Cells In Vitro. Cancers. 2019; 11(6):777. https://doi.org/10.3390/cancers11060777
Chicago/Turabian StyleJuengel, Eva, Iyad Natsheh, Ramin Najafi, Jochen Rutz, Igor Tsaur, Axel Haferkamp, Felix K.-H. Chun, and Roman A. Blaheta. 2019. "Mechanisms behind Temsirolimus Resistance Causing Reactivated Growth and Invasive Behavior of Bladder Cancer Cells In Vitro" Cancers 11, no. 6: 777. https://doi.org/10.3390/cancers11060777