Chromogranin A in the Laboratory Diagnosis of Pheochromocytoma and Paraganglioma

 , ,
, ,
Abstract
:1. Introduction
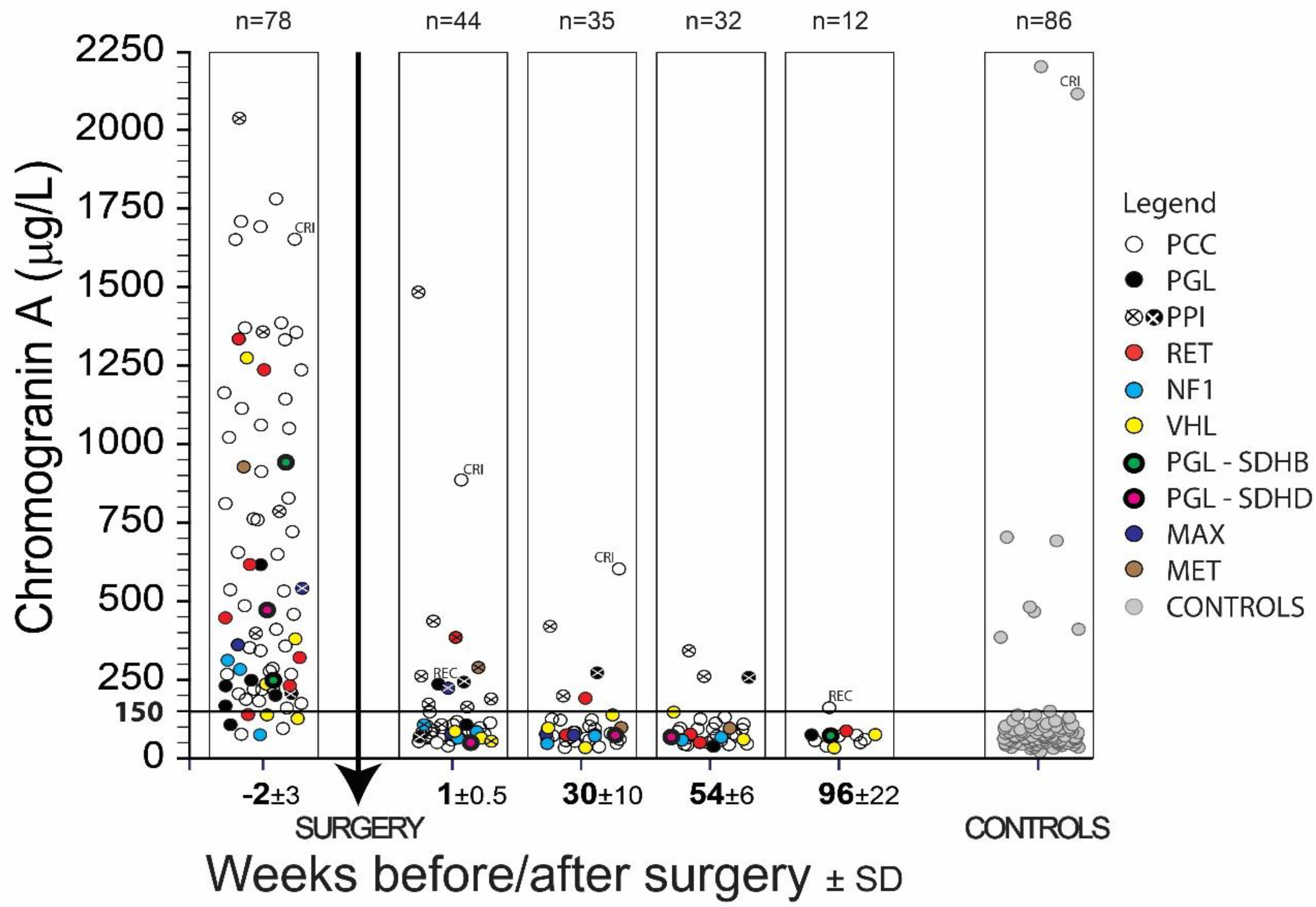
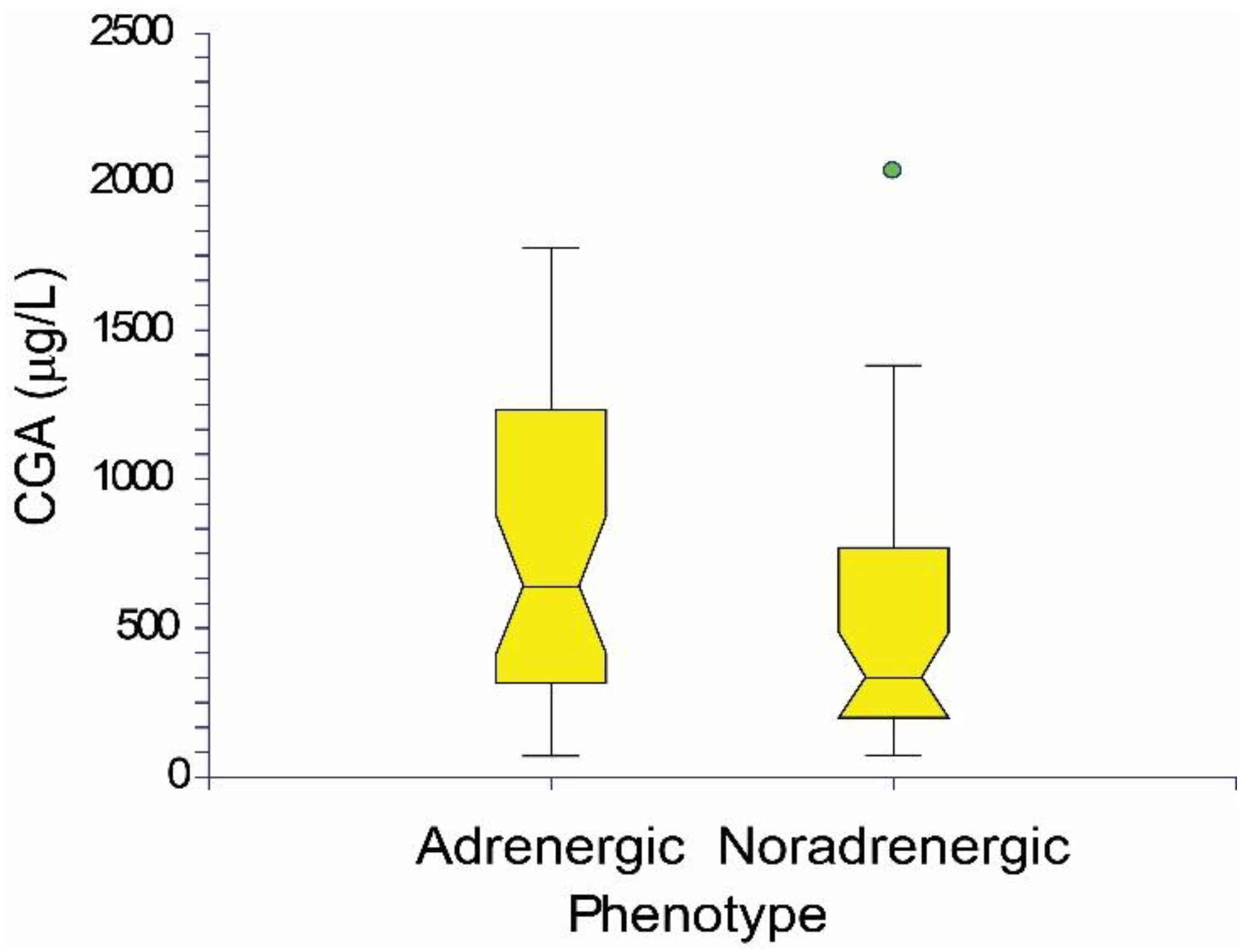
2. Results
3. Discussion
4. Materials and Methods
4.1. The Group of Patients
4.2. Laboratory Examination
4.3. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Modlin, I.M.; Gustafsson, B.I.; Moss, S.F.; Pavel, M.; Tsolakis, A.V.; Kidd, M. Chromogranin A-biological function and clinical utility in neuro endocrine tumor disease. Ann. Surg. Oncol. 2010, 17, 2427–2443. [Google Scholar] [CrossRef]
- Modlin, I.M.; Oberg, K.; Chung, D.C.; Jensen, R.T.; de Herder, W.W.; Thakker, R.V.; Caplin, M.; Delle Fave, G.; Kaltsas, G.A.; Krenning, E.P.; et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008, 9, 61–72. [Google Scholar] [CrossRef]
- Oronsky, B.; Ma, P.C.; Morgensztern, D.; Carter, C.A. Nothing but NET: A Review of Neuroendocrine Tumors and Carcinomas. Neoplasia 2017, 19, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Di Giacinto, P.; Rota, F.; Rizza, L.; Campana, D.; Isidori, A.; Lania, A.; Lenzi, A.; Zuppi, P.; Baldelli, R. Chromogranin A: From Laboratory to Clinical Aspects of Patients with Neuroendocrine Tumors. Int. J. Endocrinol. 2018, 2018, 8126087. [Google Scholar] [CrossRef]
- Marotta, V.; Zatelli, M.C.; Sciammarella, C.; Ambrosio, M.R.; Bondanelli, M.; Colao, A.; Faggiano, A. Chromogranin A as circulating marker for diagnosis and management of neuroendocrine neoplasms: More flaws than fame. Endocr. Relat. Cancer 2018, 25, R11–R29. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.S. Molecular and cellular biology of pheochromocytomas and extra-adrenal paragangliomas. Endocr. Pathol. 2006, 17, 321–328. [Google Scholar] [CrossRef]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Endocrine Society. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Pacak, K.; Tella, S.H. Pheochromocytoma and Paraganglioma. Available online: https://www.ncbi.nlm.nih.gov/books/NBK481899/ (accessed on 5 November 2018).
- Eisenhofer, G.; Lenders, J.W.; Pacak, K. Biochemical diagnosis of pheochromocytoma. Front. Horm. Res. 2004, 31, 76–106. [Google Scholar]
- Asa, S.L.; Ezzat, S.; Mete, O. The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. J. Clin. Med. 2018, 7, 280. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sippel, R.S.; O’Dorisio, M.S.; Vinik, A.I.; Lloyd, R.V.; Pacak, K. North American Neuroendocrine Tumor Society (NANETS): The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: Pheochromocytoma; paraganglioma; and medullary thyroid cancer. Pancreas 2010, 39, 775–783. [Google Scholar] [CrossRef]
- Van Berkel, A.; Lenders, J.W.; Timmers, H.J. Diagnosis of endocrine disease: Biochemical diagnosis of phaeochromocytoma and paraganglioma. Eur. J. Endocrinol. 2014, 170, R109–R119. [Google Scholar] [CrossRef]
- Andersen, K.F.; Altaf, R.; Krarup-Hansen, A.; Kromann-Andersen, B.; Horn, T.; Christensen, N.J.; Hendel, H.W. Malignant pheochromocytomas and paragangliomas—The importance of a multidisciplinary approach. Cancer Treat. Rev. 2011, 37, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Eisenhofer, G.; Mannelli, M.; Pacak, K. Phaeochromocytoma. Lancet 2005, 366, 665–675. [Google Scholar] [CrossRef]
- Crona, J.; Taieb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef] [PubMed]
- D’amico, M.A.; Ghinassi, B.; Izzicupo, P.; Manzoli, L.; Di Baldassarre, A. Biological function and clinical relevance of chromogranin A and derived peptides. Endocr. Connect. 2014, 3, R45–R54. [Google Scholar] [CrossRef] [PubMed]
- D’Herbomez, M.; Do Cao, C.; Vezzosi, D.; Borzon-Chasot, F.; Baudin, E.; Groupe des tumeurs endocrines (GTE France). Chromogranin A assay in clinical practice. Ann. Endocrinol. 2010, 71, 274–280. [Google Scholar] [CrossRef]
- Helman, L.J.; Ahn, T.G.; Levine, M.A.; Allison, A.; Cohen, P.S.; Cooper, M.J.; Cohn, D.V.; Israel, M.A. Molecular cloning and primary structure of human chromogranin A (secretory protein I) cDNA. J. Biol. Chem. 1988, 263, 11559–11563. [Google Scholar] [PubMed]
- Courel, M.; Rodemer, C.; Nguyen, S.T.; Pance, A.; Jackson, A.P.; O’Connor, D.T.; Taupenot, L. Secretory granule biogenesis in sympathoadrenal cells: Identification of a granulogenic determinant in the secretory prohormone chromogranin A. J. Biol. Chem. 2006, 281, 38038–38051. [Google Scholar] [CrossRef] [PubMed]
- Koshimizu, H.; Kim, T.; Cawley, N.X.; Loh, Y.P. Reprint of: Chromogranin A: A new proposal for trafficking; processing and induction of granule biogenesis. Regul. Pept. 2010, 165, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Marcucci, F.; Bachetti, T. Circulating chromogranin A and its fragments as diagnostic and prognostic disease markers. Pflugers Arch. 2018, 470, 199–210. [Google Scholar] [CrossRef]
- Borges, R.; Díaz-Vera, J.; Domínguez, N.; Arnau, M.R.; Machado, J.D. Chromogranins as regulators of exocytosis. J. Neurochem. 2010, 114, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H. Secretory granules in inositol 1;4;5-trisphosphate-dependent Ca2+ signaling in the cytoplasm of neuroendocrine cells. FASEB J. 2010, 24, 653–664. [Google Scholar] [CrossRef]
- Nelson, N.; Harvey, W.R. Vacuolar and plasma membrane proton-adenosinetriphosphatases. Physiol. Rev. 1999, 79, 361–385. [Google Scholar] [CrossRef]
- Kimura, N.; Takekoshi, K.; Naruse, M. Risk Stratification on Pheochromocytoma and Paraganglioma from Laboratory and Clinical Medicine. J. Clin. Med. 2018, 7, 242. [Google Scholar] [CrossRef]
- Zuber, S.; Wesley, R.; Prodanov, T.; Eisenhofer, G.; Pacak, K.; Kantorovich, V. Clinical utility of chromogranin A in SDHx-related paragangliomas. Eur. J. Clin. Investig. 2014, 44, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.D. Phaeochromocytoma of the adrenal gland scoring scale (PASS) to separate benign from malignant neoplasms. A clinicopathologic and immunophenotypic study of 100 cases. Am. J. Surg. Pathol. 2002, 26, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Szalat, A.; Fraenkel, M.; Doviner, V.; Salmon, A.; Gross, D.J. Malignant pheochromocytoma: Predictive factors of malignancy and clinical course in 16 patients at a single tertiary medical center. Endocrine 2011, 39, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Li, Z.; Cheng, C.; Yang, T.; Wang, C.; Liu, L.; Liu, S. Diagnostic value of circulating chromogranin a for neuroendocrine tumors: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0124884. [Google Scholar] [CrossRef]
- Bilek, R.; Safarik, L.; Ciprova, V.; Vlcek, P.; Lisa, L. Chromogranin A; a member of neuroendocrine secretory proteins as a selective marker for laboratory diagnosis of pheochromocytoma. Physiol. Res. 2008, 57, S171–S179. [Google Scholar]
- D’Herbomez, M.; Gouze, V.; Huglo, D.; Nocaudie, M.; Pattou, F.; Proye, C.; Wemeau, J.L.; Marchandise, X. Chromogranin A assay and (131)I-MIBG scintigraphy for diagnosis and follow-up of pheochromocytoma. J. Nucl. Med. 2001, 42, 993–997. [Google Scholar] [PubMed]
- Giovanella, L.; Squin, N.; Ghelfo, A.; Ceriani, L. Chromogranin A immunoradiometric assay in diagnosis of pheochromocytoma: Comparison with plasma metanephrines and 123I-MIBG scan. Q. J. Nucl. Med. Mol. Imaging 2006, 50, 344–347. [Google Scholar] [PubMed]
- Vinik, A.I.; Woltering, E.A.; Warner, R.R.; Caplin, M.; O’Dorisio, T.M.; Wiseman, G.A.; Coppola, D.; Go, V.L.; North American Neuroendocrine Tumor Society (NANETS). North American Neuroendocrine Tumor Society (NANETS). NANETS consensus guidelines for the diagnosis of neuroendocrine tumor. Pancreas 2010, 39, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, M.; Vacante, M.; Fichera, R.; Cappellani, A.; Cristaldi, E.; Motta, M. Chromogranin A (CgA) serum level as a marker of progression in hepatocellular carcinoma (HCC) of elderly patients. Arch. Gerontol. Geriatr. 2010, 51, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, B.; Gustafsson, B.I.; Kidd, M.; Pavel, M.; Svejda, B.; Modlin, I.M. The clinical relevance of chromogranin A as a biomarker for gastroenteropancreatic neuroendocrine tumors. Endocrinol. Metab. Clin. N. Am. 2011, 40, 111–134. [Google Scholar] [CrossRef]
- Giovinazzo, F.; Schimmack, S.; Svejda, B.; Alaimo, D.; Pfragner, R.; Modlin, I.; Kidd, M. Chromogranin A and its fragments as regulators of small intestinal neuroendocrine neoplasm proliferation. PLoS ONE 2013, 8, e81111. [Google Scholar] [CrossRef]
- Korse, C.M.; Muller, M.; Taal, B.G. Discontinuation of proton pump inhibitors during assessment of chromogranin A levels in patients with neuroendocrine tumours. Br. J. Cancer 2011, 105, 1173–1175. [Google Scholar] [CrossRef]
- Mosli, H.H.; Dennis, A.; Kocha, W.; Asher, L.J.; Van Uum, S.H. Effect of short-term proton pump inhibitor treatment and its discontinuation on chromogranin A in healthy subjects. J. Clin. Endocrinol. Metab. 2012, 97, E1731–E1735. [Google Scholar] [CrossRef]
- O’Connor, D.T.; Pandlan, M.R.; Carlton, E.; Cervenka, J.H.; Hslao, R.J. Rapid radioimmunoassay of circulating chromogranin A: In vitro stability; exploration of the neuroendocrine character of neoplasia; and assessment of the effects of organ failure. Clin. Chem. 1989, 35, 1631–1637. [Google Scholar]
- Mikkelsen, G.; Asberg, A.; Hultström, M.E.; Aasarod, K.; Hov, G.G. Reference limits for chromogranin A; CYFRA 21–1; CA 125; CA 19–9 and carcinoembryonic antigen in patients with chronic kidney disease. Int. J. Biol. Markers 2017, 32, e461–e466. [Google Scholar] [CrossRef]
- Peracchi, M.; Gebbia, C.; Basilisco, G.; Quatrini, M.; Tarantino, C.; Vescarelli, C.; Massironi, S.; Conte, D. Plasma chromogranin A in patients with autoimmune chronic atrophic gastritis; enterochromaffin-like cell lesions and gastric carcinoids. Eur. J. Endocrinol. 2005, 152, 443–448. [Google Scholar] [CrossRef]
- Massironi, S.; Fraquelli, M.; Paggi, S.; Sangiovanni, A.; Conte, D.; Sciola, V.; Ciafardini, C.; Colombo, M.; Peracchi, M. Chromogranin A levels in chronic liver disease and hepatocellular carcinoma. Dig. Liver. Dis. 2009, 41, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, R.; McAlindon, M.E.; Leeds, J.S.; Skilling, J.; Sanders, D.S. The role of serum chromogranin A in diarrhoea predominant irritable bowel syndrome. J. Gastrointestin. Liver. Dis. 2009, 18, 23–26. [Google Scholar]
- Estensen, M.E.; Hognestad, A.; Syversen, U.; Squire, I.; Ng, L.; Kjekshus, J.; Dickstein, K.; Omland, T. Prognostic value of plasma chromogranin A levels in patients with complicated myocardial infarction. Am. Heart J. 2006, 152, e1–e6. [Google Scholar] [CrossRef]
- Jansson, A.M.; Rosjo, H.; Omland, T.; Karlsson, T.; Hartford, M.; Flyvbjerg, A.; Caidahl, K. Prognostic value of circulating chromogranin A levels in acute coronary syndromes. Eur. Heart J. 2009, 30, 25–32. [Google Scholar] [CrossRef]
- Zelinka, T.; Petrak, O.; Turkova, H.; Holaj, R.; Strauch, B.; Krsek, M.; Vrankova, A.B.; Musil, Z.; Dusková, J.; Kubinyi, J.; et al. High incidence of cardiovascular complications in pheochromocytoma. Horm. Metab. Res. 2012, 44, 379–384. [Google Scholar] [CrossRef]
- Bilek, R.; Zelinka, T.; Vlcek, P.; Duskova, J.; Michalsky, D.; Novak, K.; Vaclavikova, E.; Widimsky, J., Jr. Radioimmunoassay of chromogranin A and free metanephrines in diagnosis of pheochromocytoma. Physiol. Res. 2017, 66, S397–S408. [Google Scholar]
- Eisenhofer, G.; Lenders, J.W.; Goldstein, D.S.; Mannelli, M.; Csako, G.; Walther, M.M.; Brouwers, F.M.; Pacak, K. Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin. Chem. 2005, 51, 735–744. [Google Scholar] [CrossRef]
- Van Duinen, N.; Kema, I.P.; Romijn, J.A.; Corssmit, E.P. Plasma chromogranin A levels are increased in a small portion of patients with hereditary head and neck paragangliomas. Clin. Endocrinol. 2011, 74, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Stenman, A.; Zedenius, J.; Juhlin, C.C. The Value of Histological Algorithms to Predict the Malignancy Potential of Pheochromocytomas and Abdominal Paragangliomas. A Meta-Analysis and Systematic Review of the Literature. Cancers 2019, 11, 225. [Google Scholar] [CrossRef]
- Plouin, P.F.; Amar, L.; Dekkers, O.M.; Fassnacht, M.; Gimenez-Roqueplo, A.P.; Lenders, J.W.; Lussey-Lepoutre, C.; Steichen, O.; Guideline Working Group. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur. J. Endocrinol. 2016, 174, G1–G10. [Google Scholar] [CrossRef]
- Yoo, S.H.; Albanesi, J.P. Ca2(+)-induced conformational change and aggregation of chromogranin A. J. Biol. Chem. 1990, 265, 14414–14421. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
| PPGL—Patients with Pheochromocytoma (PCC) or Paraganglioma (PGL) | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Disease | Genetics | Gender | n | 2 ± 3 Weeks before Surgery | 1 ± 0.5 Week after Surgery | 6 Months (30 ± 10 Weeks) after Surgery | 1 Year (54 ± 6 Weeks) after Surgery | 2 Years (96 ± 22 Weeks) after Surgery | ||||||||||||||||
| AGE (Years) | CGA (µg/L) | Clinical Sensitivity | n | CGA (µg/L) | Clinical Specificity | n | CGA (µg/L) | Clinical Specificity | n | CGA (µg/L) | Clinical Specificity | n | CGA (µg/L) | Clinical Specificity | ||||||||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | mean | SD | Mean | SD | |||||||||||||
| PPGL | total | total | 71 | 49 | 17 | 618.9 | 479.7 | 90 | 27 | 76 | 24.4 | 100 | 31 | 75.9 | 33.8 | 97 | 29 | 73.1 | 28.6 | 100 | 11 | 59.2 | 16.7 | 100 |
| males | 38 | 47 | 16 | 638.7 | 512 | 92 | 15 | 69.1 | 21.5 | 100 | 17 | 71.6 | 28.6 | 100 | 16 | 73.1 | 31 | 100 | 6 | 64.1 | 16.6 | 100 | ||
| females | 33 | 50 | 18 | 596.1 | 446.5 | 88 | 12 | 84.6 | 26 | 100 | 14 | 81.2 | 39.7 | 93 | 13 | 73.2 | 26.6 | 100 | 5 | 53.4 | 16.7 | 100 | ||
| PCC | total | total | 62 | 48 | 17 | 657.1 | 492.7 | 90 | 25 | 76 | 24 | 100 | 30 | 76.2 | 34.3 | 97 | 27 | 74.9 | 28.6 | 100 | 9 | 57.0 | 17.9 | 100 |
| males | 35 | 48 | 16 | 667.4 | 522.8 | 91 | 14 | 70.7 | 21.3 | 100 | 16 | 71.8 | 29.5 | 100 | 15 | 73.7 | 32 | 100 | 4 | 61.5 | 20.8 | 100 | ||
| females | 27 | 48 | 18 | 643.7 | 460.4 | 89 | 11 | 82.8 | 26.5 | 100 | 14 | 81.2 | 39.7 | 93 | 12 | 76.4 | 25 | 100 | 5 | 53.4 | 16.7 | 100 | ||
| not specified | total | 45 | 52 | 16 | 718.9 | 506 | 96 | 20 | 77.1 | 26.4 | 100 | 20 | 71.2 | 27.3 | 100 | 20 | 74.8 | 27.5 | 100 | 6 | 55.0 | 13.6 | 100 | |
| males | 25 | 52 | 15 | 758.4 | 543.4 | 100 | 10 | 70.6 | 24.6 | 100 | 11 | 70.7 | 24.8 | 100 | 10 | 70 | 30 | 100 | 3 | 54.3 | 18.2 | 100 | ||
| females | 20 | 52 | 16 | 669.6 | 464.2 | 90 | 10 | 83.7 | 27.8 | 100 | 9 | 71.8 | 31.5 | 100 | 10 | 79.6 | 25.5 | 100 | 3 | 55.7 | 11.4 | 100 | ||
| RET | total | 7 | 34 | 9 | 614.8 | 482.6 | 86 | - | - | - | - | 2 | 129 | 82.8 | 50 | 2 | 60.5 | 20 | 100 | 1 | 83.3 | - | 100 | |
| males | 3 | 42 | 6 | 693.7 | 603 | 67 | - | - | - | - | - | - | - | - | - | - | - | - | 1 | 83.3 | - | 100 | ||
| females | 4 | 28 | 5 | 555.6 | 460.9 | 100 | - | - | - | - | 2 | 129 | 82.8 | 50 | 2 | 60.5 | 20 | 100 | - | - | - | - | ||
| VHL | total | 5 | 32 | 21 | 427.9 | 482.5 | 60 | 2 | 70.9 | 14.9 | 100 | 3 | 85 | 52.2 | 100 | 2 | 99.1 | 62.6 | 100 | 2 | 49.9 | 28.5 | 100 | |
| males | 3 | 22 | 2 | 244.2 | 127.4 | 67 | 2 | 70.9 | 14.9 | 100 | 2 | 82.3 | 73.5 | 100 | 2 | 99.1 | 62.6 | 100 | - | - | - | - | ||
| females | 2 | 46 | 34 | 703.6 | 803.5 | 50 | - | - | - | 100 | 1 | 90.3 | - | 100 | - | - | - | - | 2 | 49.9 | 28.5 | 100 | ||
| NF1 | total | 3 | 54 | 10 | 220.2 | 128.8 | 67 | 2 | 71.4 | 15.4 | 100 | 2 | 56.2 | 16.9 | 100 | 2 | 57.8 | 7.5 | 100 | - | - | - | - | |
| males | 3 | 54 | 10 | 220.2 | 128.8 | 67 | 2 | 71.4 | 15.4 | 100 | 2 | 56.2 | 16.9 | 100 | 2 | 57.8 | 7.5 | 100 | - | - | - | - | ||
| females | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| MAX | total | 1 | 60 | - | 357.9 | - | 100 | 1 | 74.2 | - | 100 | 2 | 71.5 | 2.1 | 100 | - | - | - | - | - | - | - | - | |
| males | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| females | 1 | 60 | - | 357.9 | - | 100 | 1 | 74.2 | - | 100 | 2 | 71.5 | 2.1 | 100 | - | - | - | - | - | - | - | - | ||
| MET | total | 1 | 47 | - | 925 | - | 100 | - | - | - | - | 1 | 94.4 | - | 100 | 1 | 92 | - | 100 | - | - | - | - | |
| males | 1 | 47 | - | 925 | - | 100 | - | - | - | - | 1 | 94.4 | - | 100 | 1 | 92 | - | 100 | - | - | - | - | ||
| females | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| PGL | total | total | 9 | 52 | 20 | 355.9 | 270.8 | 89 | 2 | 74.9 | 40.4 | 100 | 1 | 67.3 | - | 100 | 2 | 49.3 | 20.7 | 100 | 2 | 69.1 | 2.8 | 100 |
| males | 3 | 36 | 12 | 303.7 | 145.1 | 100 | 1 | 46.4 | - | 100 | 1 | 67.3 | - | 100 | 1 | 64 | - | 100 | 2 | 69.1 | 2.8 | 100 | ||
| females | 6 | 61 | 18 | 382 | 326.3 | 83 | 1 | 103.5 | - | 100 | - | - | - | - | 1 | 34.6 | - | 100 | - | - | - | - | ||
| not specified | total | 6 | 56 | 22 | 258.2 | 181.1 | 83 | 1 | 103.5 | - | 100 | - | - | - | - | 1 | 34.6 | - | 100 | 1 | 71.1 | - | 100 | |
| males | 1 | 32 | - | 197.1 | - | 100 | - | - | - | - | - | - | - | - | - | - | - | - | 1 | 71.1 | - | 100 | ||
| females | 5 | 61 | 20 | 270.5 | 199.7 | 80 | 1 | 103.5 | - | 100 | - | - | - | - | 1 | 34.6 | - | 100 | - | - | - | - | ||
| SDHB | total | 2 | 55 | 7 | 592.2 | 190.9 | 100 | - | - | - | - | - | - | - | - | - | - | - | - | 1 | 67.1 | - | 100 | |
| males | 1 | 50 | - | 245.1 | - | 100 | - | - | - | - | - | - | - | - | - | - | - | - | 1 | 67.1 | - | 100 | ||
| females | 1 | 60 | - | 939.4 | - | 100 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| SDHD | total | 1 | 26 | - | 469 | - | 100 | 1 | 46.4 | - | 100 | 1 | 67.3 | - | 100 | 1 | 64 | - | 100 | - | - | - | - | |
| males | 1 | 26 | - | 469 | - | 100 | 1 | 46.4 | - | 100 | 1 | 67.3 | - | 100 | 1 | 64 | - | 100 | - | - | - | - | ||
| females | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Controls | |||||||
|---|---|---|---|---|---|---|---|
| Syndrome | Gender | n | AGE (years) | CGA µg/L) | Clinical Specificity | ||
| Mean | SD | Mean | SD | ||||
| total | total | 85 | 53 | 19 | 125.3 | 259.3 | 92 |
| males | 29 | 53 | 21 | 102.6 | 128.9 | 93 | |
| females | 56 | 54 | 18 | 134 | 306.3 | 91 | |
| adrenocortical adenoma | total | 44 | 63 | 11 | 178.5 | 352.7 | 84 |
| males | 15 | 65 | 6 | 135.2 | 173.3 | 87 | |
| females | 29 | 62 | 13 | 200.9 | 417.7 | 83 | |
| adrenocortical adenoma + MTC, FTC | total | 1 | 71 | - | 31.8 | - | 100 |
| males | - | - | - | - | - | - | |
| females | 1 | 71 | - | 31.8 | - | 100 | |
| adrenocortical adenoma + PTC | total | 1 | 75 | - | 108.4 | - | 100 |
| males | - | - | - | - | - | - | |
| females | 1 | 75 | - | 108.4 | - | 100 | |
| hypertension | total | 8 | 45 | 16 | 43.2 | 11 | 100 |
| males | 4 | 41 | 22 | 50.2 | 9.1 | 100 | |
| females | 4 | 48 | 8 | 36.3 | 8.3 | 100 | |
| MEN 2A | total | 2 | 10 | 4 | 70.6 | 15.2 | 100 |
| males | 1 | 13 | - | 81.3 | - | 100 | |
| females | 1 | 7 | - | 59.8 | - | 100 | |
| MEN 2A + MTC | total | 7 | 26 | 14 | 69.5 | 35.5 | 100 |
| males | 3 | 20 | 12 | 71.8 | 24.9 | 100 | |
| females | 4 | 30 | 16 | 67.7 | 45.7 | 100 | |
| MEN 2B + MTC | total | 3 | 30 | 16 | 90.7 | 53.9 | 100 |
| males | 1 | 17 | - | 146.2 | - | 100 | |
| females | 2 | 37 | 16 | 62.9 | 34.4 | 100 | |
| MTC | total | 9 | 56 | 13 | 76.9 | 36.2 | 100 |
| males | 2 | 51 | 17 | 51.3 | 10.9 | 100 | |
| females | 7 | 57 | 13 | 84.2 | 38.1 | 100 | |
| MTC + PTC | total | 1 | 70 | - | 42.3 | - | 100 |
| males | 1 | 70 | - | 42.3 | - | 100 | |
| females | - | - | - | - | - | - | |
| PTC | total | 4 | 58 | 22 | 81.1 | 40.4 | 100 |
| males | 1 | 80 | - | 102.6 | - | 100 | |
| females | 3 | 50 | 20 | 73.9 | 46.2 | 100 | |
| thyroid disorders | total | 5 | 36 | 18 | 70.2 | 12.2 | 100 |
| males | 1 | 51 | - | 54.6 | - | 100 | |
| females | 4 | 32 | 18 | 74.1 | 9.9 | 100 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bílek, R.; Vlček, P.; Šafařík, L.; Michalský, D.; Novák, K.; Dušková, J.; Václavíková, E.; Widimský, J., Jr.; Zelinka, T. Chromogranin A in the Laboratory Diagnosis of Pheochromocytoma and Paraganglioma. Cancers 2019, 11, 586. https://doi.org/10.3390/cancers11040586
Bílek R, Vlček P, Šafařík L, Michalský D, Novák K, Dušková J, Václavíková E, Widimský J Jr., Zelinka T. Chromogranin A in the Laboratory Diagnosis of Pheochromocytoma and Paraganglioma. Cancers. 2019; 11(4):586. https://doi.org/10.3390/cancers11040586
Chicago/Turabian StyleBílek, Radovan, Petr Vlček, Libor Šafařík, David Michalský, Květoslav Novák, Jaroslava Dušková, Eliška Václavíková, Jiří Widimský, Jr., and Tomáš Zelinka. 2019. "Chromogranin A in the Laboratory Diagnosis of Pheochromocytoma and Paraganglioma" Cancers 11, no. 4: 586. https://doi.org/10.3390/cancers11040586