The Role of Platelets in Cancer Pathophysiology: Focus on Malignant Glioma


Abstract
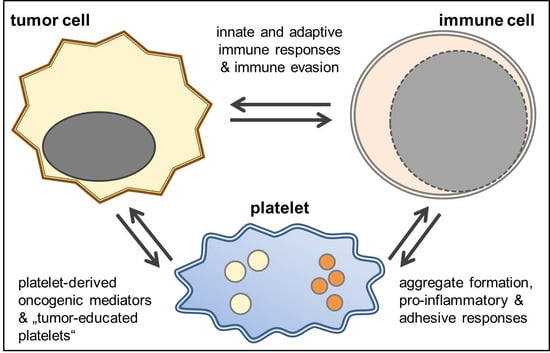
:
{kind=link}
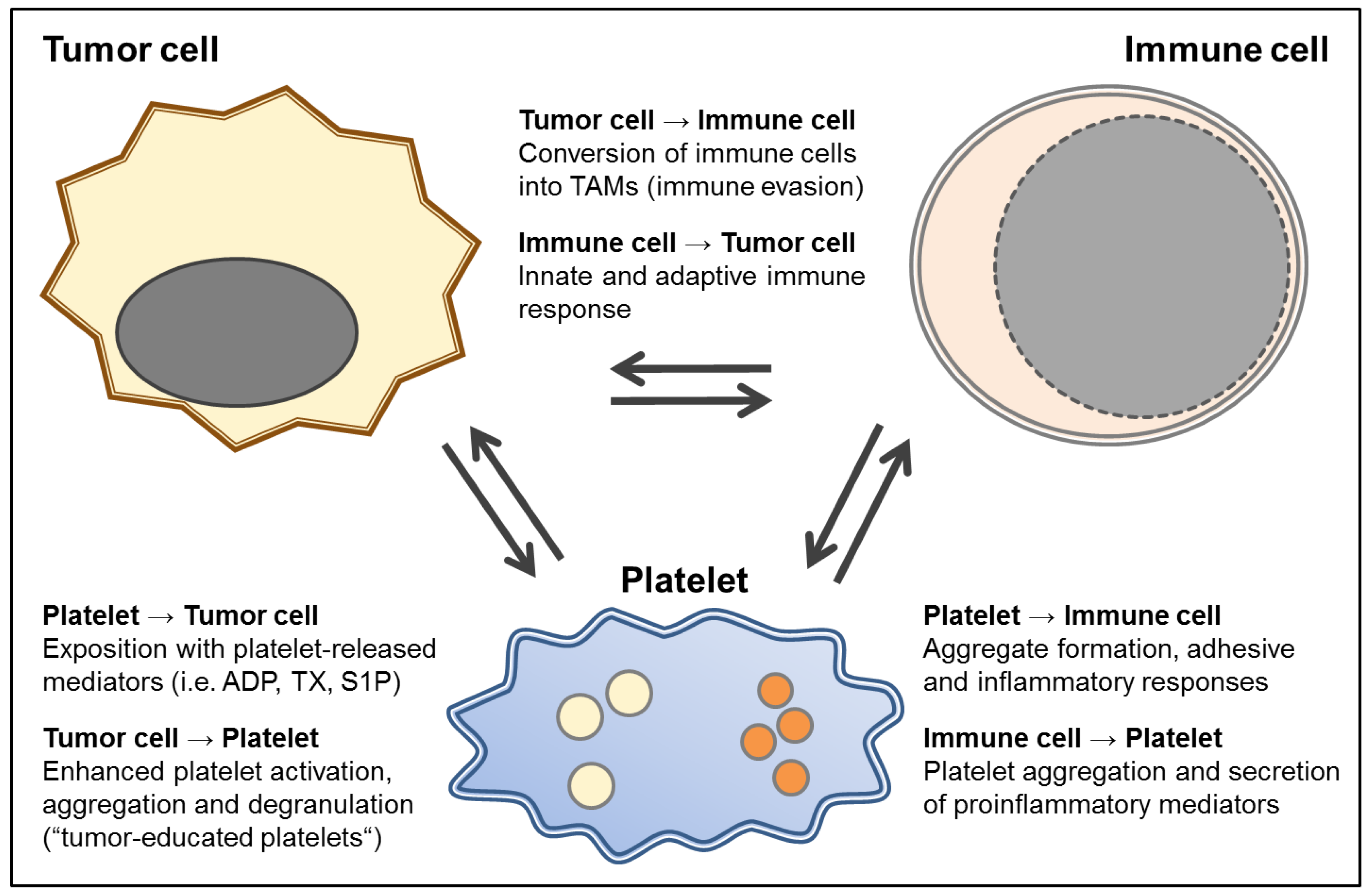{kind=link}
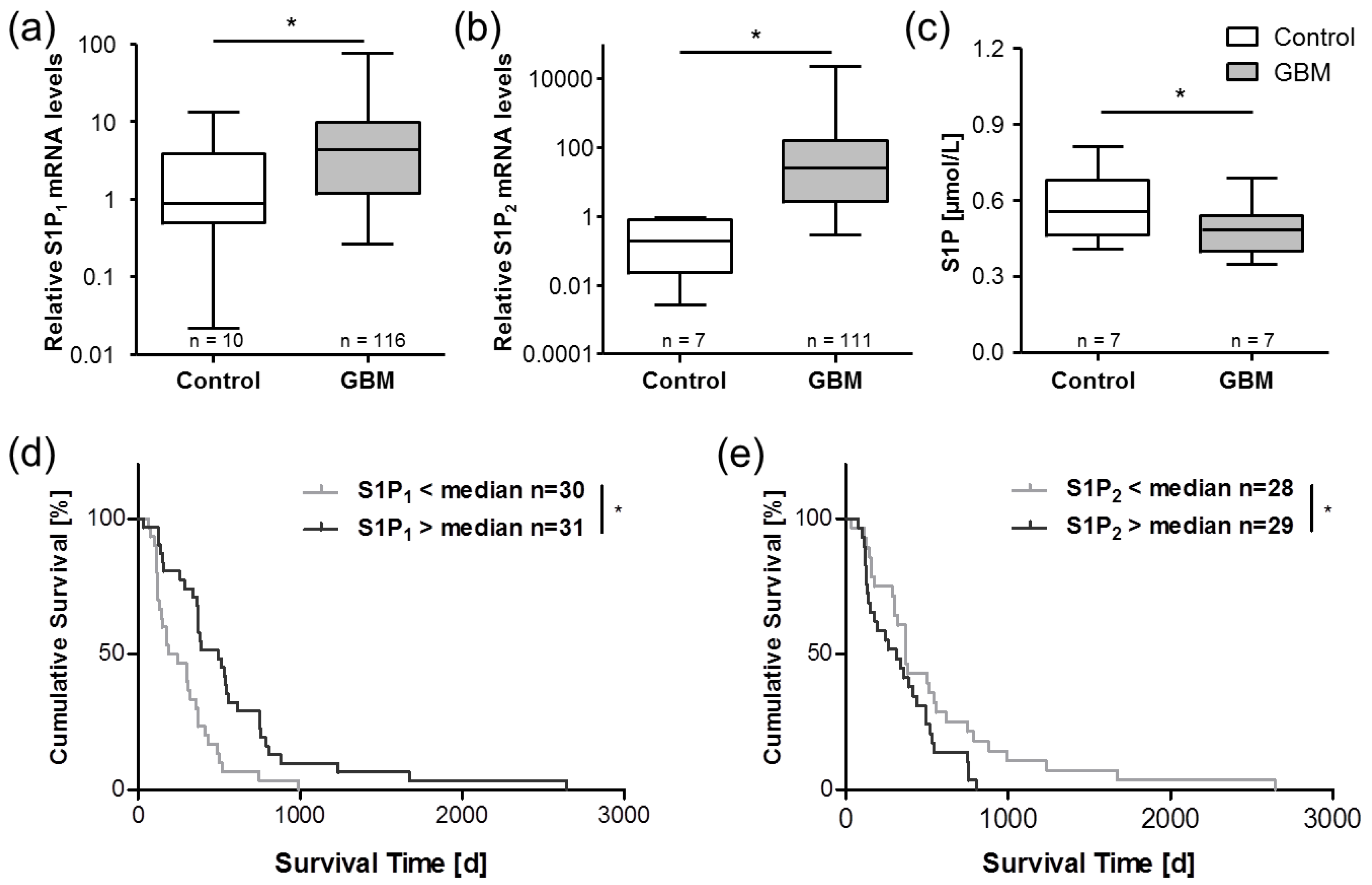{kind=link}
1. Introduction
2. Platelets and Solid Tumors
3. Platelets and Glioblastoma
4. The Immunomodulatory Potential of Platelets in GBM
5. Sphingosine-1-Phosphate in Glioblastoma
6. Summary and Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy with Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Lim, M. Immunotherapy for Glioblastoma: Playing Chess, Not Checkers. Clin. Cancer Res. 2018, 24, 4059–4061. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Reardon, D.A.; Wucherpfennig, K.; Chiocca, E.A. Immunotherapy for glioblastoma: On the sidelines or in the game? Discov. Med. 2017, 24, 201–208. [Google Scholar] [PubMed]
- Sayegh, E.T.; Oh, T.; Fakurnejad, S.; Bloch, O.; Parsa, A.T. Vaccine therapies for patients with glioblastoma. J. Neurooncol. 2014, 119, 531–546. [Google Scholar] [CrossRef]
- Schijns, V.; Pretto, C.; Strik, A.M.; Gloudemans-Rijkers, R.; Deviller, L.; Pierre, D.; Chung, J.; Dandekar, M.; Carrillo, J.A.; Kong, X.T.; et al. Therapeutic Immunization against Glioblastoma. Int. J. Mol. Sci. 2018, 19, 2540. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Verla, T.; Sanchez-Perez, L.; Reap, E.A.; Choi, B.D.; Fecci, P.E.; Sampson, J.H. Immunotherapy for malignant glioma. Surg. Neurol. Int. 2015, 6, S68–S77. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kalin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef]
- Bowman, R.L.; Joyce, J.A. Therapeutic targeting of tumor-associated macrophages and microglia in glioblastoma. Immunotherapy 2014, 6, 663–666. [Google Scholar] [CrossRef]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006, 8, 261–279. [Google Scholar] [CrossRef]
- Parney, I.F.; Waldron, J.S.; Parsa, A.T. Flow cytometry and in vitro analysis of human glioma-associated macrophages. Laboratory investigation. J. Neurosurg. 2009, 110, 572–582. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; Rath, D.; Gawaz, M. Role of chemokine receptors CXCR4 and CXCR7 for platelet function. Biochem. Soc. Trans. 2015, 43, 720–726. [Google Scholar] [CrossRef]
- Stellos, K.; Gawaz, M. Platelets and stromal cell-derived factor-1 in progenitor cell recruitment. Semin. Thromb. Hemost. 2007, 33, 159–164. [Google Scholar] [CrossRef]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Sierko, E.; Hempel, D.; Tucker, S.C.; Honn, K.V. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017, 36, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Huong, P.T.; Nguyen, L.T.; Nguyen, X.B.; Lee, S.K.; Bach, D.H. The Role of Platelets in the Tumor-Microenvironment and the Drug Resistance of Cancer Cells. Cancers 2019, 11, 240. [Google Scholar] [CrossRef]
- Navone, S.E.; Guarnaccia, L.; Locatelli, M.; Rampini, P.; Caroli, M.; la Verde, N.; Gaudino, C.; Bettinardi, N.; Riboni, L.; Marfia, G.; et al. Significance and Prognostic Value of The Coagulation Profile in Patients with Glioblastoma: Implications for Personalized Therapy. World Neurosurg. 2019, 121, e621–e629. [Google Scholar] [CrossRef]
- Fisher, J.L.; Palmisano, S.; Schwartzbaum, J.A.; Svensson, T.; Lonn, S. Comorbid conditions associated with glioblastoma. J. Neurooncol. 2014, 116, 585–591. [Google Scholar] [CrossRef]
- Smith, T.R.; Lall, R.R.; Graham, R.B.; McClendon, J.; Lall, R.R.; Nanney, A.D.; Adel, J.G.; Zakarija, A.; Chandler, J.P. Venous thromboembolism in high grade glioma among surgical patients: Results from a single center over a 10 year period. J. Neurooncol. 2014, 120, 347–352. [Google Scholar] [CrossRef]
- Streiff, M.B.; Ye, X.; Kickler, T.S.; Desideri, S.; Jani, J.; Fisher, J.; Grossman, S.A. A prospective multicenter study of venous thromboembolism in patients with newly-diagnosed high-grade glioma: Hazard rate and risk factors. J. Neurooncol. 2015, 124, 299–305. [Google Scholar] [CrossRef]
- Rong, Y.; Durden, D.L.; van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’ necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef]
- Edwin, N.C.; Khoury, M.N.; Sohal, D.; McCrae, K.R.; Ahluwalia, M.S.; Khorana, A.A. Recurrent venous thromboembolism in glioblastoma. Thromb. Res. 2016, 137, 184–188. [Google Scholar] [CrossRef]
- Brat, D.J.; van Meir, E.G. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Investig. 2004, 84, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Simanek, R.; Vormittag, R.; Hassler, M.; Roessler, K.; Schwarz, M.; Zielinski, C.; Pabinger, I.; Marosi, C. Venous thromboembolism and survival in patients with high-grade glioma. Neuro Oncol. 2007, 9, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Meikle, C.K.; Kelly, C.A.; Garg, P.; Wuescher, L.M.; Ali, R.A.; Worth, R.G. Cancer and Thrombosis: The Platelet Perspective. Front Cell Dev. Biol. 2016, 4, 147. [Google Scholar] [CrossRef]
- Buergy, D.; Wenz, F.; Groden, C.; Brockmann, M.A. Tumor-platelet interaction in solid tumors. Int. J. Cancer 2012, 130, 2747–2760. [Google Scholar] [CrossRef] [Green Version]
- Riess, L. Zur pathologischen Anatomie des Blutes. Arch. Anat. Physiol. Wissensch. Med. 1872, 39, 237–249. [Google Scholar]
- Billroth, T. Lectures on Surgical Pathology and Therapeutics: A Handbook for Students and Practitionersed; The New Sydenham Society: London, UK, 1878. [Google Scholar]
- Heinmoller, E.; Weinel, R.J.; Heidtmann, H.H.; Salge, U.; Seitz, R.; Schmitz, I.; Muller, K.M.; Zirngibl, H. Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. J. Cancer Res. Clin. Oncol. 1996, 122, 735–744. [Google Scholar] [CrossRef]
- Jacobs, E.J.; Newton, C.C.; Gapstur, S.M.; Thun, M.J. Daily aspirin use and cancer mortality in a large US cohort. J. Natl. Cancer Inst. 2012, 104, 1208–1217. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Schrör, K.; Rauch, B.H. Aspirin and lipid mediators in the cardiovascular system. Prostaglandins Other Lipid Mediat. 2015, 121, 17–23. [Google Scholar] [CrossRef]
- Riedl, J.; Kaider, A.; Marosi, C.; Prager, G.W.; Eichelberger, B.; Assinger, A.; Pabinger, I.; Panzer, S.; Ay, C. Decreased platelet reactivity in patients with cancer is associated with high risk of venous thromboembolism and poor prognosis. Thromb. Haemost. 2017, 117, 90–98. [Google Scholar] [CrossRef]
- Placke, T.; Kopp, H.G.; Salih, H.R. Modulation of natural killer cell anti-tumor reactivity by platelets. J. Innate Immun. 2011, 3, 374–382. [Google Scholar] [CrossRef]
- Zhang, W.; Dang, S.; Hong, T.; Tang, J.; Fan, J.; Bu, D.; Sun, Y.; Wang, Z.; Wisniewski, T. A humanized single-chain antibody against beta 3 integrin inhibits pulmonary metastasis by preferentially fragmenting activated platelets in the tumor microenvironment. Blood 2012, 120, 2889–2898. [Google Scholar] [CrossRef] [Green Version]
- Borsig, L.; Wong, R.; Feramisco, J.; Nadeau, D.R.; Varki, N.M.; Varki, A. Heparin and cancer revisited: Mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc. Natl. Acad. Sci. USA 2001, 98, 3352–3357. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.J.; Balaj, L.; Hulleman, E.; van Rijn, S.; Pegtel, D.M.; Walraven, M.; Widmark, A.; Gerritsen, W.R.; Verheul, H.M.; Vandertop, W.P.; et al. Blood platelets contain tumor-derived RNA biomarkers. Blood 2011, 118, 3680–3683. [Google Scholar] [CrossRef] [Green Version]
- In’t Veld, S.; Sjors, G.J.G.; Wurdinger, T. Tumor-educated platelets. Blood 2019. [Google Scholar] [CrossRef]
- Olsson, A.K.; Cedervall, J. The pro-inflammatory role of platelets in cancer. Platelets 2018, 29, 569–573. [Google Scholar] [CrossRef]
- Koenen, R.R. The prowess of platelets in immunity and inflammation. Thromb. Haemost. 2016, 116, 605–612. [Google Scholar] [CrossRef]
- Lam, F.W.; Vijayan, K.V.; Rumbaut, R.E. Platelets and Their Interactions with Other Immune Cells. Compr. Physiol. 2015, 5, 1265–1280. [Google Scholar]
- Mahajan-Thakur, S.; Böhm, A.; Jedlitschky, G.; Schrör, K.; Rauch, B.H. Sphingosine-1-Phosphate and Its Receptors: A Mutual Link between Blood Coagulation and Inflammation. Mediat. Inflamm. 2015, 2015, 831059. [Google Scholar] [CrossRef]
- Mahajan-Thakur, S.; Sostmann, B.D.; Fender, A.C.; Behrendt, D.; Felix, S.B.; Schrör, K.; Rauch, B.H. Sphingosine-1-phosphate induces thrombin receptor PAR-4 expression to enhance cell migration and COX-2 formation in human monocytes. J. Leukoc. Biol. 2014, 96, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Aleman, M.M.; Gardiner, C.; Harrison, P.; Wolberg, A.S. Differential contributions of monocyte- and platelet-derived microparticles towards thrombin generation and fibrin formation and stability. J. Thromb. Haemost. 2011, 9, 2251–2261. [Google Scholar] [CrossRef]
- Brockmann, M.A.; Giese, A.; Mueller, K.; Kaba, F.J.; Lohr, F.; Weiss, C.; Gottschalk, S.; Nolte, I.; Leppert, J.; Tuettenberg, J.; et al. Preoperative thrombocytosis predicts poor survival in patients with glioblastoma. Neuro Oncol. 2007, 9, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, M.; Carvalho, B.; Vaz, R.; Linhares, P. Influence of neutrophil-lymphocyte ratio in prognosis of glioblastoma multiforme. J. Neurooncol. 2018, 136, 173–180. [Google Scholar] [CrossRef]
- Karsy, M.; Gelbman, M.; Shah, P.; Balumbu, O.; Moy, F.; Arslan, E. Established and emerging variants of glioblastoma multiforme: Review of morphological and molecular features. Folia Neuropathol. 2012, 50, 301–321. [Google Scholar] [CrossRef]
- Karsy, M.; Huang, T.; Kleinman, G.; Karpel-Massler, G. Molecular, histopathological, and genomic variants of glioblastoma. Front Biosci. 2014, 19, 1065–1087. [Google Scholar] [CrossRef]
- Marx, S.; Splittstöhser, M.; Kinnen, F.; Moritz, E.; Joseph, C.; Paul, S.; Paland, H.; Seifert, C.; Marx, M.; Böhm, A.; et al. Platelet activation parameters and platelet-leucocyte-conjugate formation in glioblastoma multiforme patients. Oncotarget 2018, 9, 25860–25876. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, S.E.; Moore, S.C.; Pfeiffer, R.M.; Inskip, P.D.; Park, Y.; Hollenbeck, A.; Rajaraman, P. Nonsteroidal anti-inflammatory drugs and glioma in the NIH-AARP Diet and Health Study cohort. Cancer Prev. Res. 2011, 4, 2027–2034. [Google Scholar] [CrossRef]
- Gaist, D.; Garcia-Rodriguez, L.A.; Sorensen, H.T.; Hallas, J.; Friis, S. Use of low-dose aspirin and non-aspirin nonsteroidal anti-inflammatory drugs and risk of glioma: A case-control study. Br. J. Cancer 2013, 108, 1189–1194. [Google Scholar] [CrossRef]
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar] [Green Version]
- Peerschke, E.I.; Yin, W.; Ghebrehiwet, B. Complement activation on platelets: Implications for vascular inflammation and thrombosis. Mol. Immunol. 2010, 47, 2170–2175. [Google Scholar] [CrossRef]
- Lösche, W.; Heptinstall, S. Value of Platelet Activation Markers as Prothrombotic Risk Indicators. Transfus Med. Hemother. 2007, 34, 34–42. [Google Scholar] [CrossRef]
- Zarbock, A.; Polanowska-Grabowska, R.K.; Ley, K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev. 2007, 21, 99–111. [Google Scholar] [CrossRef]
- Storey, R.F.; James, S.K.; Siegbahn, A.; Varenhorst, C.; Held, C.; Ycas, J.; Husted, S.E.; Cannon, C.P.; Becker, R.C.; Steg, P.G.; et al. Lower mortality following pulmonary adverse events and sepsis with ticagrelor compared to clopidogrel in the PLATO study. Platelets 2014, 25, 517–525. [Google Scholar] [CrossRef]
- Coppinger, J.A.; O’Connor, R.; Wynne, K.; Flanagan, M.; Sullivan, M.; Maguire, P.B.; Fitzgerald, D.J.; Cagney, G. Moderation of the platelet releasate response by aspirin. Blood 2007, 109, 4786–4792. [Google Scholar] [CrossRef] [Green Version]
- Blair, P.; Flaumenhaft, R. Platelet alpha-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef]
- Scheuerer, B.; Ernst, M.; Durrbaum-Landmann, I.; Fleischer, J.; Grage-Griebenow, E.; Brandt, E.; Flad, H.D.; Petersen, F. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood 2000, 95, 1158–1166. [Google Scholar]
- Loppnow, H.; Libby, P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. J. Clin. Investig. 1990, 85, 731–738. [Google Scholar] [CrossRef]
- Kopp, H.G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef]
- Lee, Y.L.; Lee, L.W.; Su, C.Y.; Hsiao, G.; Yang, Y.Y.; Leu, S.J.; Shieh, Y.H.; Burnouf, T. Virally inactivated human platelet concentrate lysate induces regulatory T cells and immunosuppressive effect in a murine asthma model. Transfusion 2013, 53, 1918–1928. [Google Scholar] [CrossRef]
- Sengelov, L.; Kamby, C.; Schou, G.; von der Maase, H. Prognostic factors and significance of chemotherapy in patients with recurrent or metastatic transitional cell cancer of the urinary tract. Cancer 1994, 74, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Panek, W.K.; Pituch, K.C.; Miska, J.; Kim, J.W.; Rashidi, A.; Kanojia, D.; Lopez-Rosas, A.; Han, Y.; Yu, D.; Chang, C.L.; et al. Local Application of Autologous Platelet-Rich Fibrin Patch (PRF-P) Suppresses Regulatory T Cell Recruitment in a Murine Glioma Model. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mahajan-Thakur, S.; Bien-Moller, S.; Marx, S.; Schroeder, H.; Rauch, B.H. Sphingosine 1-phosphate (S1P) signaling in glioblastoma multiforme—A systematic review. Int. J. Mol. Sci. 2017, 18, 2448. [Google Scholar] [CrossRef]
- Pyne, S.; Pyne, N.J. New perspectives on the role of sphingosine 1-phosphate in cancer. Handb. Exp. Pharm. 2013, 216, 55–71. [Google Scholar]
- Pyne, N.J.; Ohotski, J.; Bittman, R.; Pyne, S. The role of sphingosine 1-phosphate in inflammation and cancer. Adv. Biol. Regul. 2014, 54, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bien-Moller, S.; Lange, S.; Holm, T.; Bohm, A.; Paland, H.; Kupper, J.; Herzog, S.; Weitmann, K.; Havemann, C.; Vogelgesang, S.; et al. Expression of S1P metabolizing enzymes and receptors correlate with survival time and regulate cell migration in glioblastoma multiforme. Oncotarget 2016, 7, 13031–13046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef]
- Anelli, V.; Gault, C.R.; Cheng, A.B.; Obeid, L.M. Sphingosine kinase 1 is up-regulated during hypoxia in U87MG glioma cells. Role of hypoxia-inducible factors 1 and 2. J. Biol. Chem. 2008, 283, 3365–3375. [Google Scholar] [CrossRef] [PubMed]
- Riccitelli, E.; Giussani, P.; di Vito, C.; Condomitti, G.; Tringali, C.; Caroli, M.; Galli, R.; Viani, P.; Riboni, L. Extracellular sphingosine-1-phosphate: A novel actor in human glioblastoma stem cell survival. PLoS ONE 2013, 8, e68229. [Google Scholar] [CrossRef]
- Muller, J.; von Bernstorff, W.; Heidecke, C.D.; Schulze, T. Differential S1P Receptor Profiles on M1- and M2-Polarized Macrophages Affect Macrophage Cytokine Production and Migration. Biomed. Res. Int. 2017, 2017, 7584621. [Google Scholar] [CrossRef]
- Ulrych, T.; Böhm, A.; Polzin, A.; Daum, G.; Nusing, R.M.; Geisslinger, G.; Hohlfeld, T.; Schrör, K.; Rauch, B.H. Release of sphingosine-1-phosphate from human platelets is dependent on thromboxane formation. J. Thromb. Haemost. 2011, 9, 790–798. [Google Scholar] [CrossRef] [Green Version]
- Böhm, A.; Flösser, A.; Ermler, S.; Fender, A.C.; Luth, A.; Kleuser, B.; Schrör, K.; Rauch, B.H. Factor-Xa-induced mitogenesis and migration require sphingosine kinase activity and S1P formation in human vascular smooth muscle cells. Cardiovasc. Res. 2013, 99, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Obinata, H.; Hla, T. Sphingosine 1-phosphate in coagulation and inflammation. Semin. Immunopathol. 2012, 34, 73–91. [Google Scholar] [CrossRef]
- Rauch, B.H. Sphingosine 1-phosphate as a link between blood coagulation and inflammation. Cell Physiol. Biochem. 2014, 34, 185–196. [Google Scholar] [CrossRef]
- Olivera, A.; Allende, M.L.; Proia, R.L. Shaping the landscape: Metabolic regulation of S1P gradients. Biochim. Biophys. Acta 2013, 1831, 193–202. [Google Scholar] [CrossRef]
- Heffernan-Stroud, L.A.; Obeid, L.M. Sphingosine kinase 1 in cancer. Adv. Cancer Res. 2013, 117, 201–235. [Google Scholar] [PubMed]
- Estrada-Bernal, A.; Palanichamy, K.; Chaudhury, A.R.; van Brocklyn, J.R. Induction of brain tumor stem cell apoptosis by FTY720: A potential therapeutic agent for glioblastoma. Neuro Oncol. 2012, 14, 405–415. [Google Scholar] [CrossRef]
- Cattaneo, M.G.; Vanetti, C.; Samarani, M.; Aureli, M.; Bassi, R.; Sonnino, S.; Giussani, P. Cross-talk between sphingosine-1-phosphate and EGFR signaling pathways enhances human glioblastoma cell invasiveness. Febs Lett. 2018, 592, 949–961. [Google Scholar] [CrossRef]
- Quint, K.; Stiel, N.; Neureiter, D.; Schlicker, H.U.; Nimsky, C.; Ocker, M.; Strik, H.; Kolodziej, M.A. The role of sphingosine kinase isoforms and receptors S1P1, S1P2, S1P3, and S1P5 in primary, secondary, and recurrent glioblastomas. Tumour Biol. 2014, 35, 8979–8989. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marx, S.; Xiao, Y.; Baschin, M.; Splittstöhser, M.; Altmann, R.; Moritz, E.; Jedlitschky, G.; Bien-Möller, S.; Schroeder, H.W.S.; Rauch, B.H. The Role of Platelets in Cancer Pathophysiology: Focus on Malignant Glioma. Cancers 2019, 11, 569. https://doi.org/10.3390/cancers11040569
Marx S, Xiao Y, Baschin M, Splittstöhser M, Altmann R, Moritz E, Jedlitschky G, Bien-Möller S, Schroeder HWS, Rauch BH. The Role of Platelets in Cancer Pathophysiology: Focus on Malignant Glioma. Cancers. 2019; 11(4):569. https://doi.org/10.3390/cancers11040569
Chicago/Turabian StyleMarx, Sascha, Yong Xiao, Marcel Baschin, Maximilian Splittstöhser, Robert Altmann, Eileen Moritz, Gabriele Jedlitschky, Sandra Bien-Möller, Henry W.S. Schroeder, and Bernhard H. Rauch. 2019. "The Role of Platelets in Cancer Pathophysiology: Focus on Malignant Glioma" Cancers 11, no. 4: 569. https://doi.org/10.3390/cancers11040569