Dysregulation of Macropinocytosis Processes in Glioblastomas May Be Exploited to Increase Intracellular Anti-Cancer Drug Levels: The Example of Temozolomide

 , ,
, ,
Abstract
:1. Introduction
2. Results
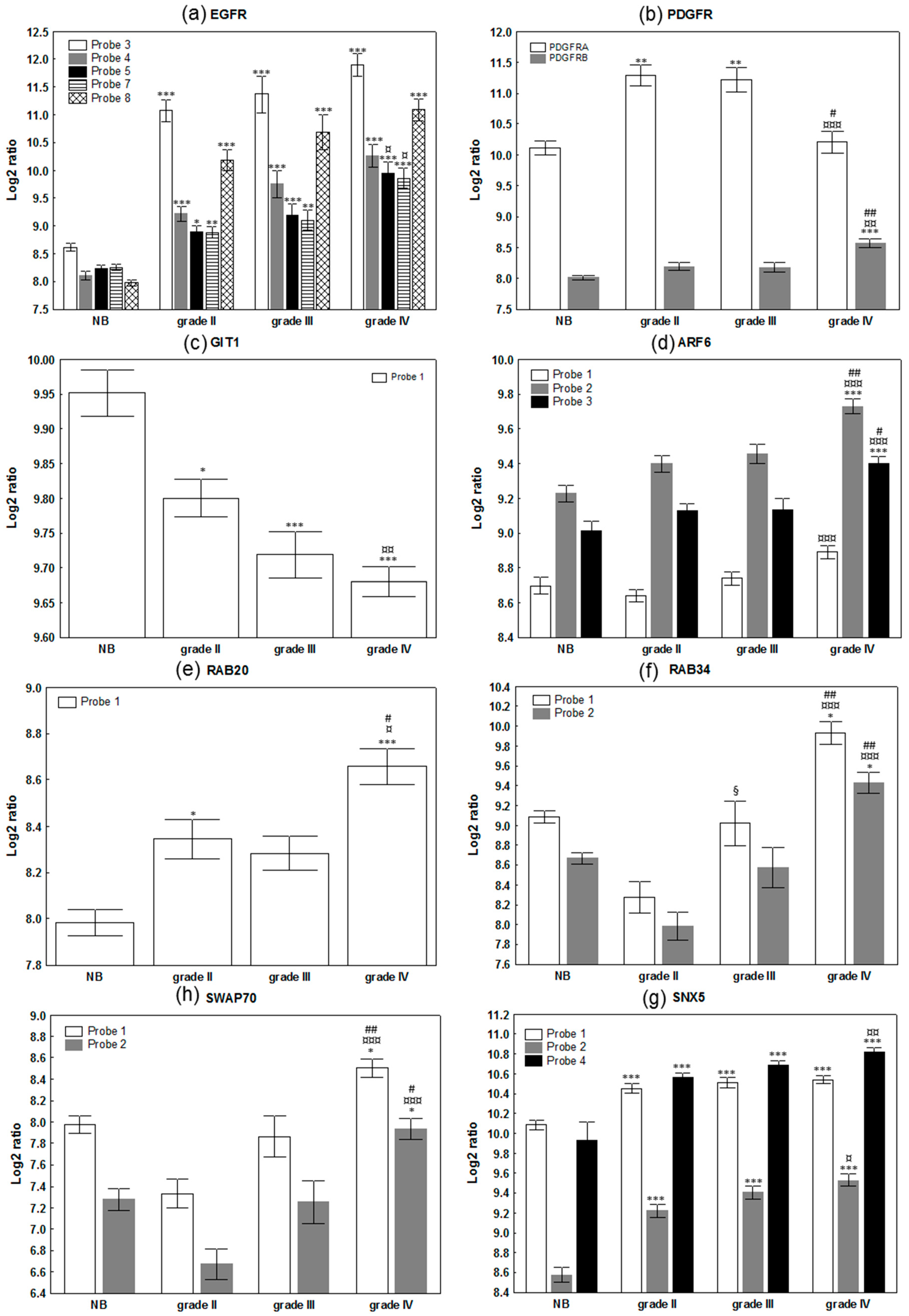
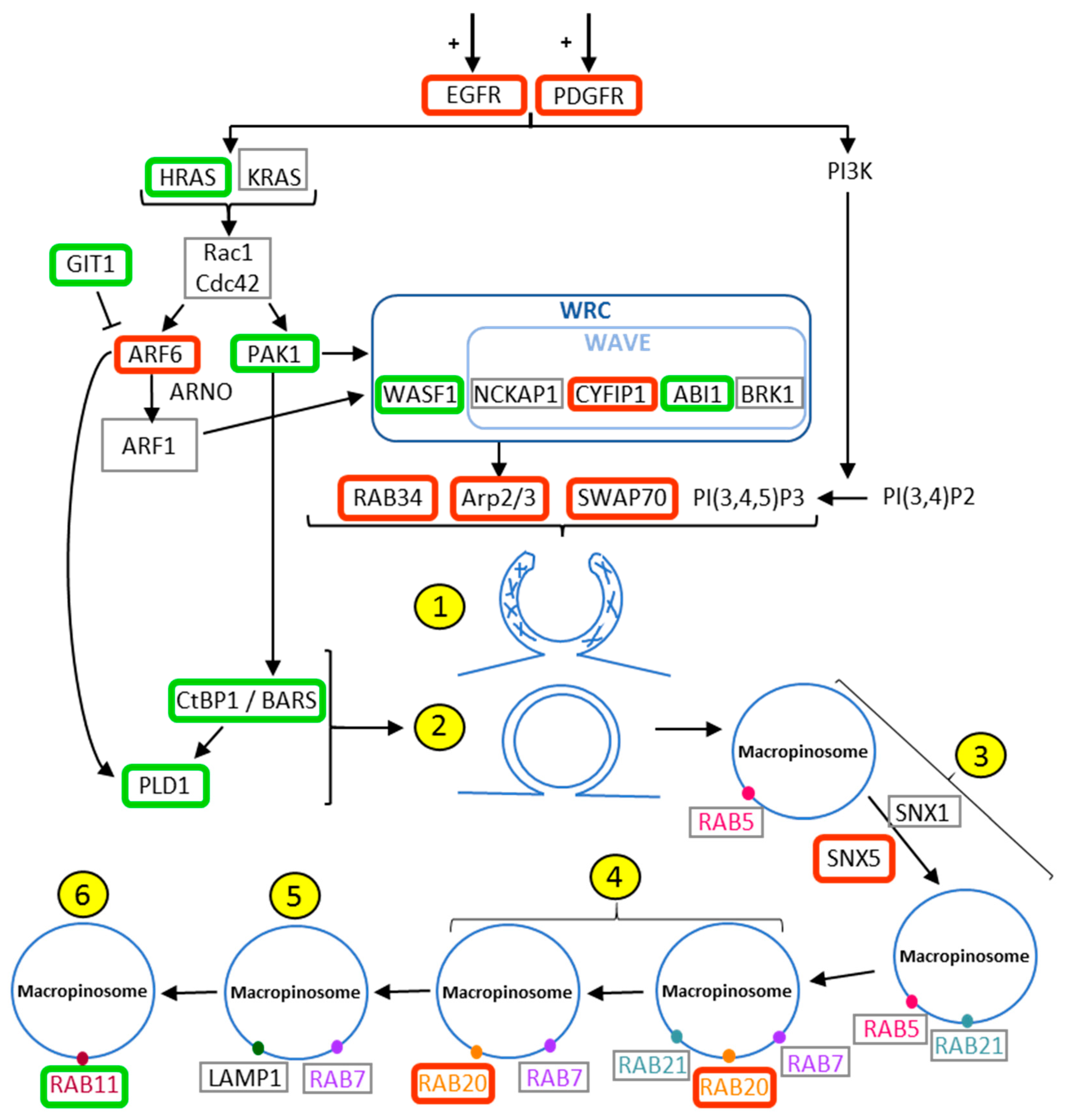
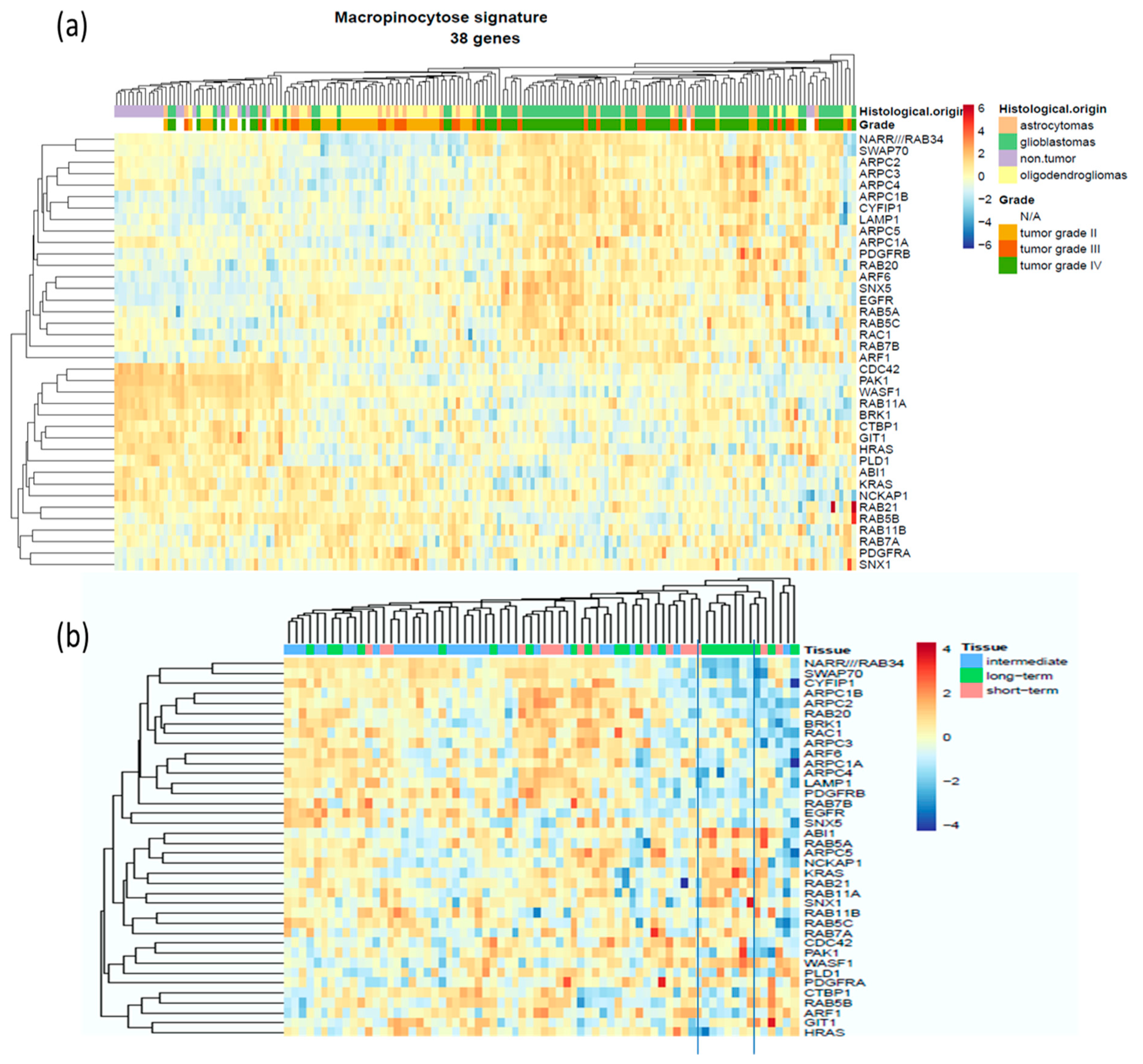
2.1. Evaluation of The Deregulation of Macropinocytosis in Human Gliomas
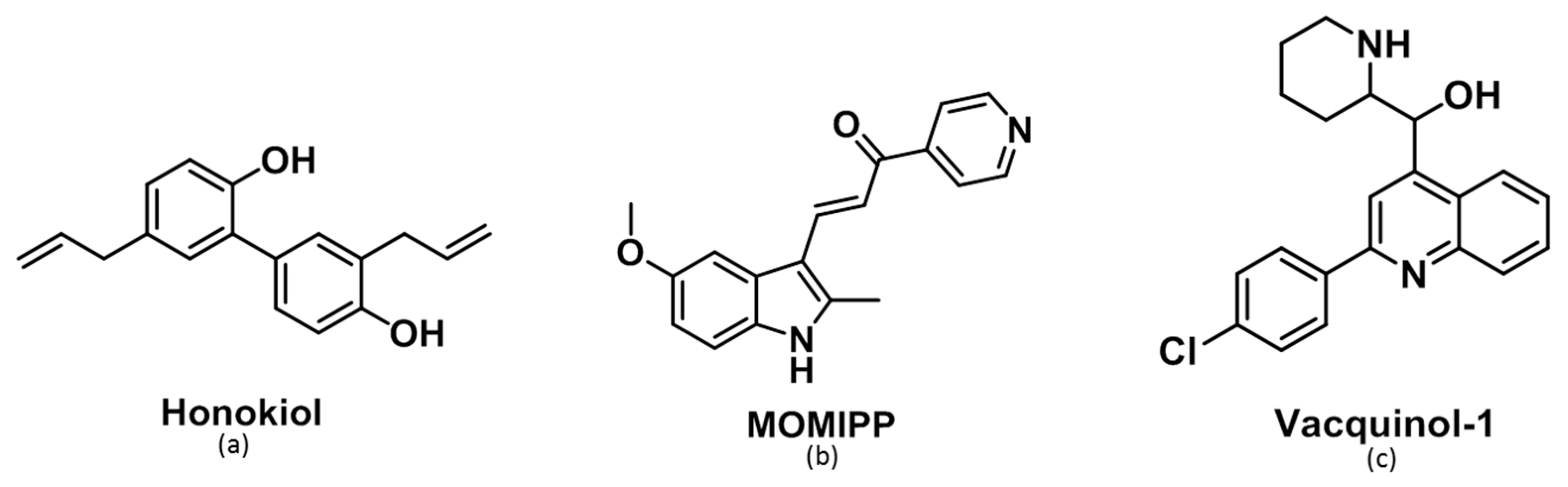
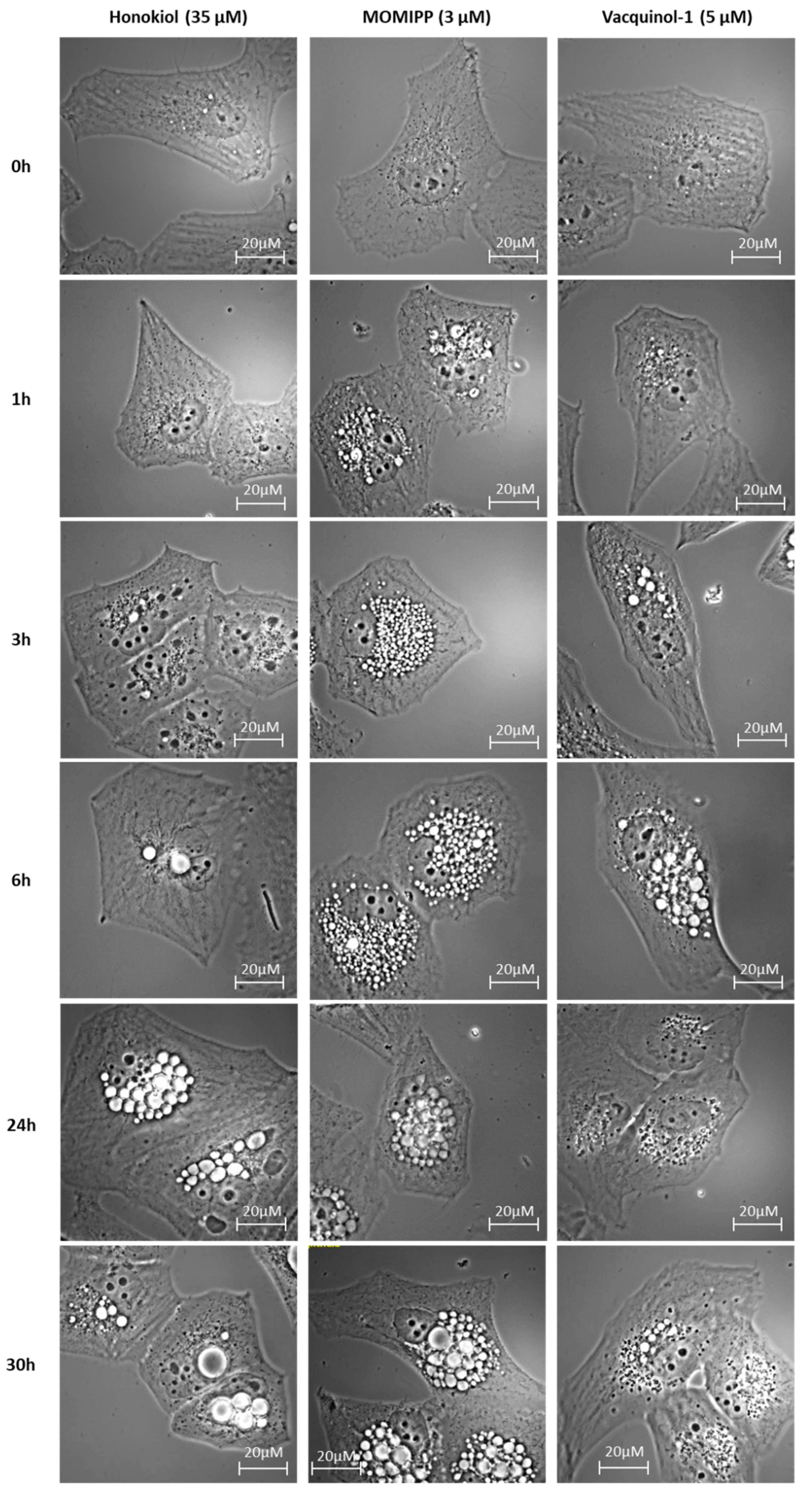
2.2. Morphological Comparison of Glioma Cells Treated by Honokiol, 3-(5-methoxy -2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (MOMIPP) and Vacquinol-1
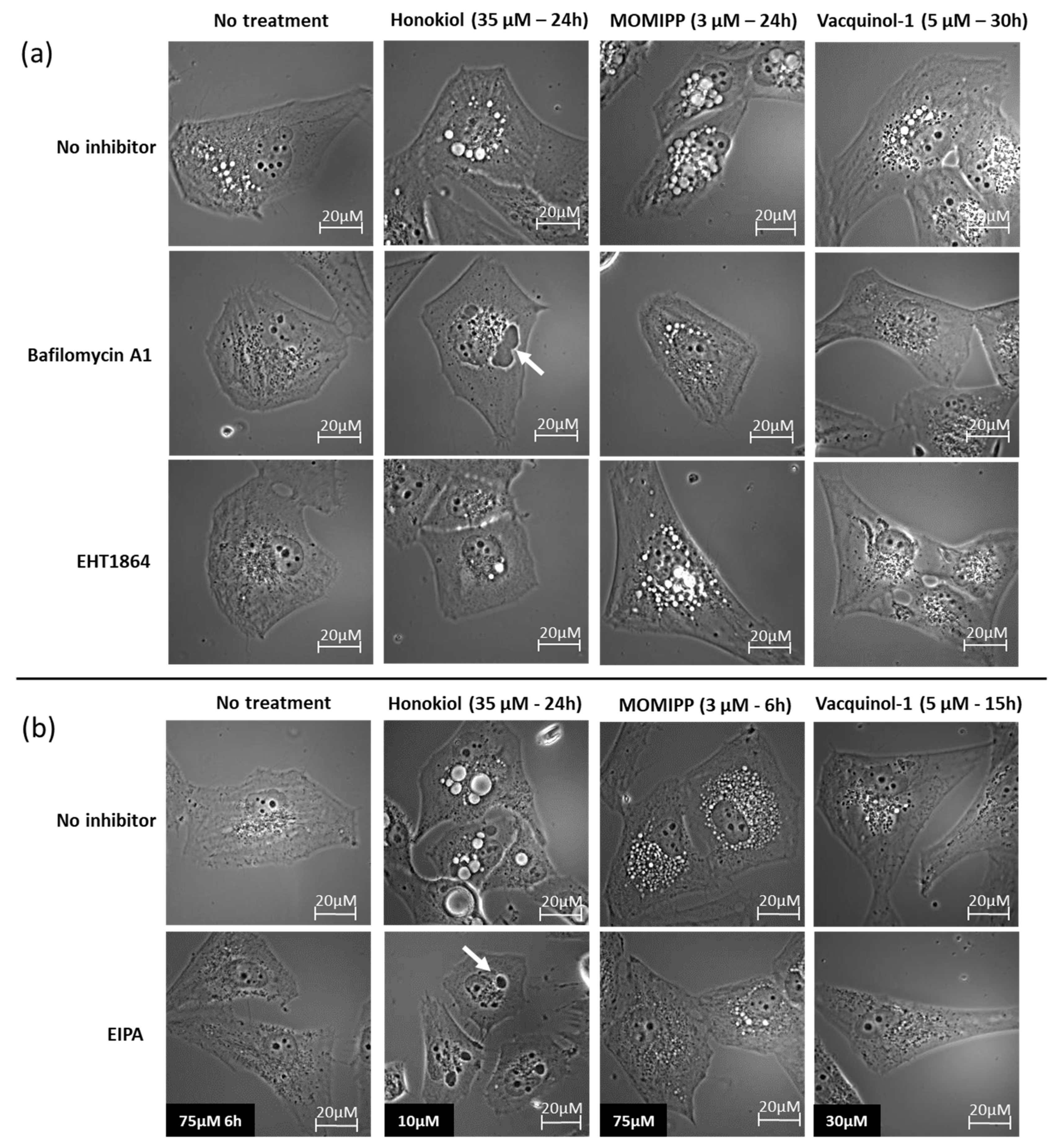
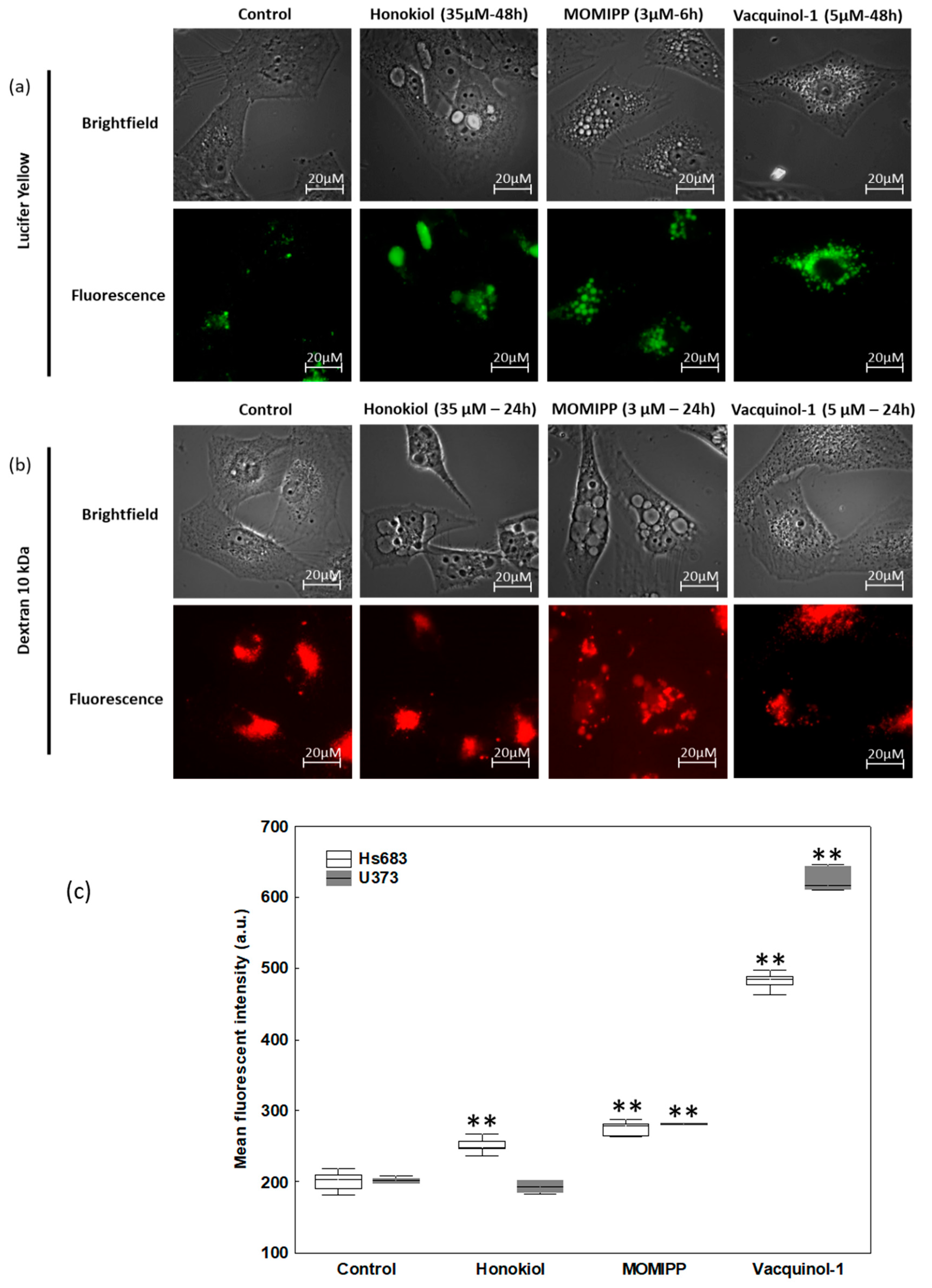
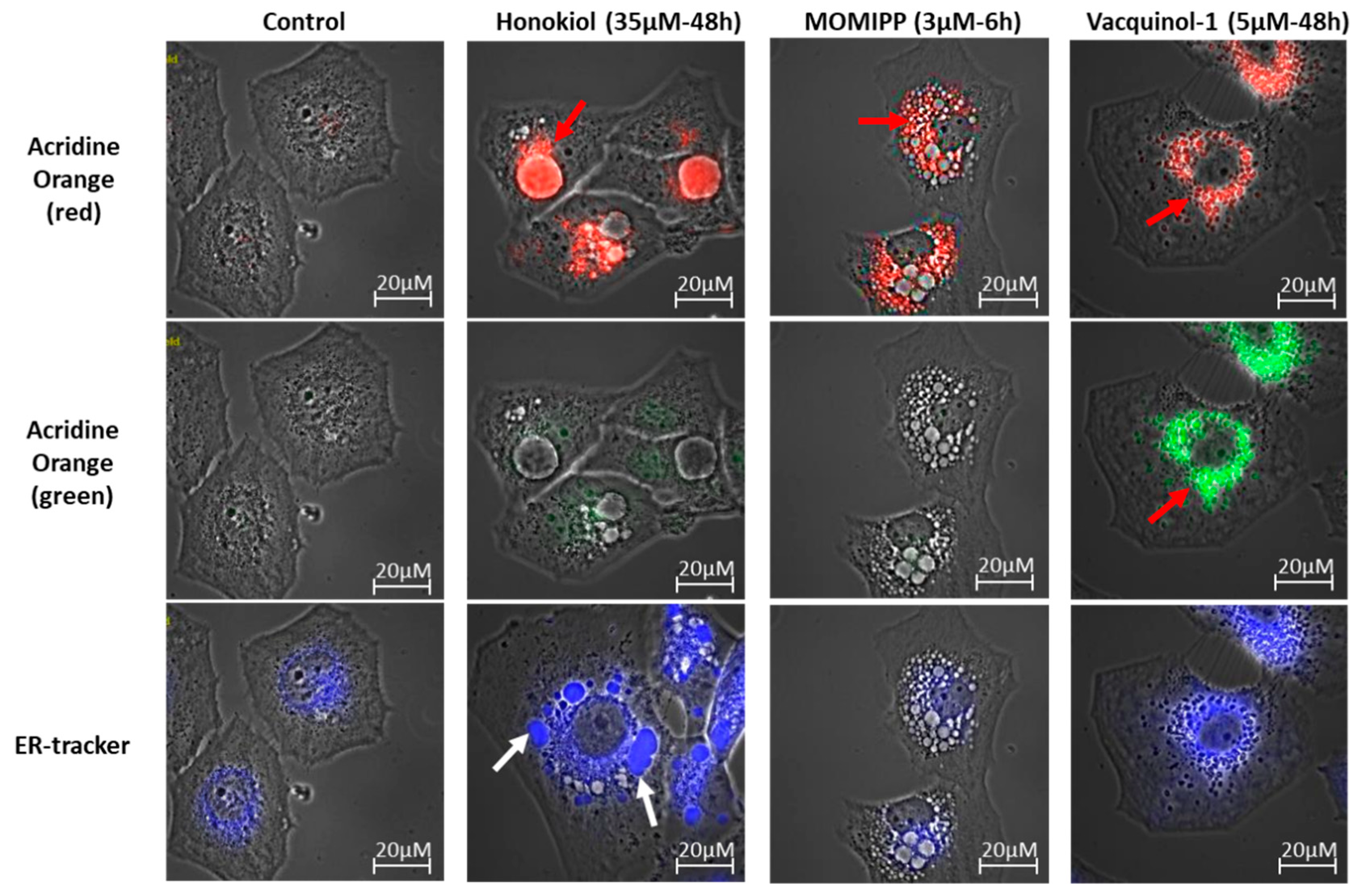
2.3. Characterization of the Vacuoles Induced by Honokiol, Vacquinol-1 and MOMIPP
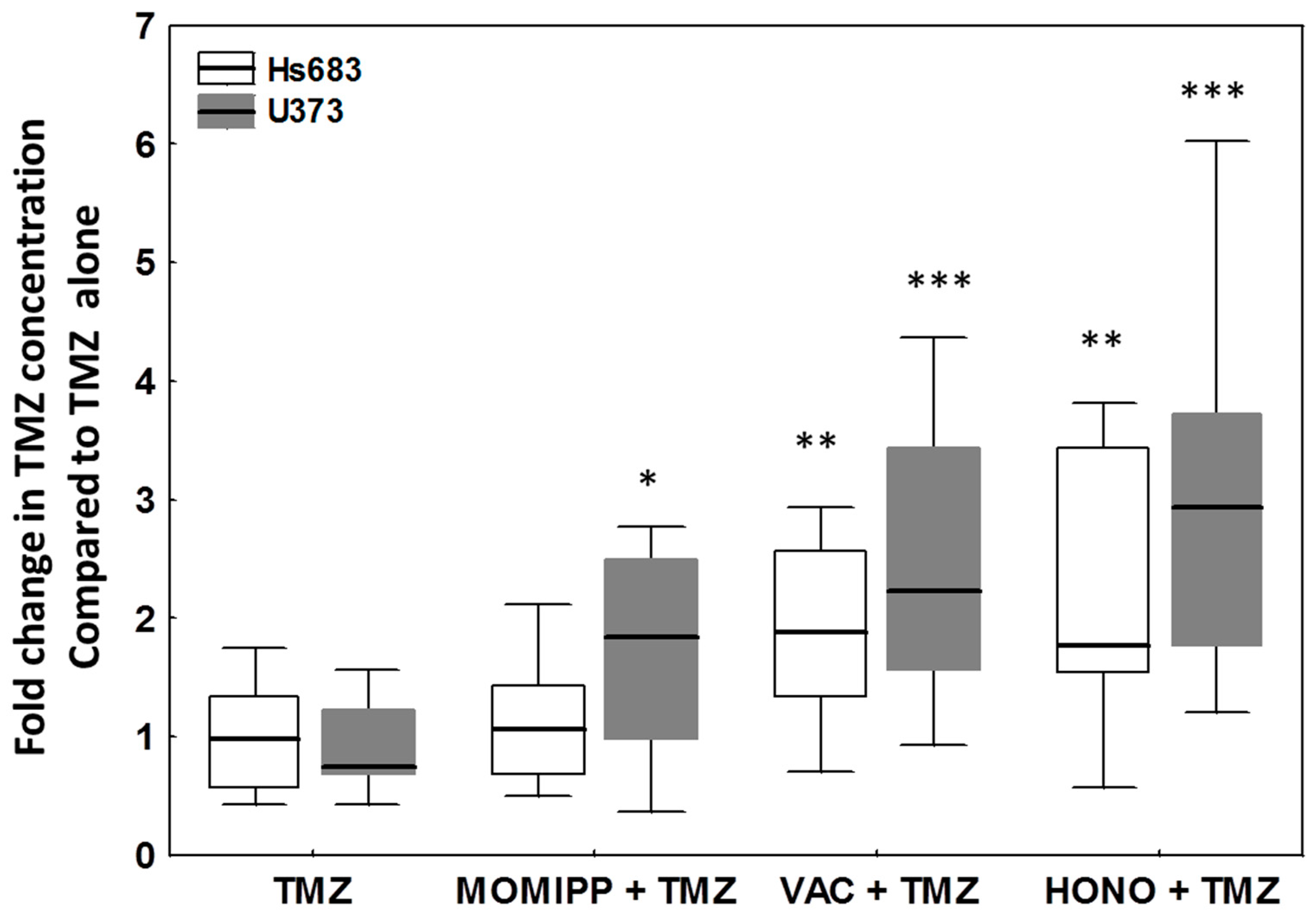
2.4. Evaluation of the Effects of Honokiol, Vacquinol-1 and MOMIPP on Intracellular Temozolomide (TMZ) Concentration
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Compounds
4.2. MTT Colorimetric Assay
4.3. Characterization of Vacuoles
4.3.1. Phase Contrast Microscopy for Morphological Observations
4.3.2. Fluorescent Microscopy Assays
4.3.3. Flow Cytometry
4.4. Quantification of Intracellular TMZ
4.4.1. Sample Preparation
4.4.2. LC-MS Process, Data Acquisition and Analysis
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Name | Number of Probe | Kruskall - Wallis | 2 Tailed | Up or Down Regulation | Role in Macropinocytosis |
|---|---|---|---|---|---|---|
| EGFR | Epidermal growth factor receptor | 8 | */**/***/***/***/***/***/*** | 1/2/3/3/4/1/4/3 | Up |
|
| PDGFRA | Platelet derived growth factor receptor alpha | 1 | *** | 4 | Up | |
| PDGFRB | Platelet derived growth factor receptor beta | 1 | *** | 3 | Up | |
| H-RAS | H-Ras proto-oncogene, GTPase | 1 | *** | 3 | Down |
|
| K-RAS | K-RAS proto-oncogene, GTPase | 5 | NS/NS/***/NS/NS | 0/0/3/0/1 | - | |
| CDC42 | Cell division cycle 42 | 5 | ***/***/***/***/*** | 2/3/4/4/3 | ? |
|
| RAC1 | Ras-related C3 botulinum toxin substrate 1 (rho family, small GTP binding protein Rac1) | 3 | ***/**/***/NS | 1/1/2/0 | - | |
| GIT1 | GIT ArfGAP 1 | 1 | *** | 4 | Down |
|
| ARF1 | ADP ribosylation factor 1 | 3 | ***/***/* | 4/1/1 | ? | |
| ARF6 | ADP ribosylation factor 6 | 3 | ***/***/*** | 1/3/3 | Up | |
| PAK1 | p21 (RAC1) activated kinase 1 | 4 | **/***/***/*** | 3/3/3/3 | Down |
|
| WASF1 | WAS protein family member 1 | 1 | *** | 4 | Down |
|
| CYFIP1 | Cytoplasmic FMR1 interacting protein 1 | 1 | *** | 3 | Up |
|
| NCKAP1 | NCK associated protein 1 | 2 | ***/NS | 2/0 | - | |
| ABI1 | Abl-interactor 1 | 2 | ***/*** | 2/4 | Down | |
| BRK1 | BRICK1, SCAR/WAVE actin nucleating complex subunit | 1 | NS | 0 | - | |
| ARPC1A | Actin related protein 2/3 complex subunit 1A | 1 | *** | 2 | - | |
| ARPC1B | Actin related protein 2/3 complex subunit 1B | 1 | *** | 4 | Up | |
| ARPC2 | Actin related protein 2/3 complex subunit 2 | 3 | ***/***/*** | 1/1/1 | Up | |
| ARPC3 | Actin related protein 2/3 complex subunit 3 | 1 | *** | 1 | Up | |
| ARPC4 | Actin related protein 2/3 complex subunit 4 | 2 | */*** | 1/1 | - | |
| ARPC5 | Actin related protein 2/3 complex subunit 5 | 4 | ***/**/***/** | 3/1/3/2 | Up | |
| SWAP70 | SWAP switching B-cell complex 70kDa subunit | 2 | ***/*** | 3/3 | Up |
|
| RAB34 | RAB34, member RAS oncogene family | 2 | ***/*** | 4/3 | Up |
|
| CTBP1 | C-terminal binding protein 1 | 4 | ***/***/NS | 3/2/1 | Down |
|
| PLD1 | Phospholipase D1 | 6 | ***/***/**/***/***/*** | 3/2/2/4/3/3 | Down | |
| RAB5A | RAB5A, member RAS oncogene family | 3 | NS/NS/*** | 0/0/4 | - |
|
| RAB5B | RAB5B, member RAS oncogene family | 1 | *** | 3 | Down | |
| RAB5C | RAB5C, member RAS oncogene family | 2 | NS/** | 0/1 | Up | |
| RAB20 | RAB20, member RAS oncogene family | 1 | *** | 4 | Up | |
| RAB21 | RAB21, member RAS oncogene family | 4 | */**/*/** | 1/2/2/2 | ? | |
| RAB7A | RAB7A, member RAS oncogene family | 6 | **/NS/***/NS/NS/* | 2/0/2/0/0/1 | - |
|
| RAB7B | RAB7B, member RAS oncogene family | 2 | *** | 1 | - | |
| LAMP1 | Lysosomal associated membrane protein 1 | 3 | */NS/NS | 1/0/0 | - |
|
| SNX1 | Sorting nexin 1 | 3 | NS/***/NS | 0/3/0 | ? |
|
| SNX5 | Sorting nexin 5 | 5 | ***/***/***/***/*** | 3/4/4/4/3 | Up | |
| RAB11A | RAB11A, member RAS oncogene family | 3 | ***/NS/*** | 2/0/4 | Down |
|
| RAB11B | RAB11B, member RAS oncogene family | 2 | */NS | 1/0 | - |
References
- Eisele, G.; Weller, M. Targeting apoptosis pathways in glioblastoma. Cancer Lett. 2013, 332, 335–345. [Google Scholar] [CrossRef]
- Lefranc, F.; Le Rhun, E.; Kiss, R.; Weller, M. Glioblastoma quo vadis: Will migration and invasiveness reemerge as therapeutic targets? Cancer Treat. Rev. 2018, 68, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 28, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Kornienko, A.; Mathieu, V.; Rastogi, S.K.; Lefranc, F.; Kiss, R. Therapeutic agents triggering nonapoptotic cancer cell death. J. Med. Chem. 2013, 56, 4823–4839. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.G.; Garside, R.; Rogers, G.; Stein, K.; Grant, R. Temozolomide for high grade glioma. Cochrane Database Syst. Rev. 2013, 4, CD007415. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.H. Pinocytosis. Bull. Johns Hopkins Hops. 1931, 49, 17–26. [Google Scholar]
- Kerr, M.C.; Teasdale, R.D. Defining macropinocytosis. Traffic 2009, 10, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Maltese, W.A.; Overmeyer, J.H. Non-apoptotic cell death associated with perturbations of macropinocytosis. Front. Physiol. 2015, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Redelman-Sidi, G.; Binyamin, A.; Gaeta, I.; Palm, W.; Thompson, C.B.; Romesser, P.B.; Lowe, S.W.; Bagul, M.; Doench, J.G.; Root, D.E.; et al. The Canonical Wnt Pathway Drives Macropinocytosis in Cancer. Cancer Res. 2018, 78, 4658–4670. [Google Scholar] [CrossRef] [PubMed]
- Overmeyer, J.H.; Kaul, A.; Johnson, E.E.; Maltese, W.A. Active Ras Triggers Death in Glioblastoma Cells through Hyperstimulation of Macropinocytosis. Mol. Cancer Res. 2008, 6, 965–977. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Wang, X.; Liu, Y.; Li, Y.; Colvin, R.A.; Tong, L.; Wu, S.; Chen, X. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett. 2014, 351, 242–251. [Google Scholar] [CrossRef]
- Veithen, A.; Cupers, P.; Baudhuin, P.; Courtoy, P.J. v-Src induces constitutive macropinocytosis in rat fibroblasts. J. Cell Sci. 1996, 109, 2005–2012. [Google Scholar]
- Kasahara, K.; Nakayama, Y.; Sato, I.; Ikeda, K.; Hoshino, M.; Endo, T.; Yamaguchi, N. Role of Src-Family Kinases in Formation and Trafficking of Macropinosomes. J. Cell. Physiol. 2007, 211, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Kodaz, H.; Kostek, O.; Hacioglu, M.B.; Erdogan, B.; Kodaz, C.E.; Hacibekiroglu, I.; Turkmen, E.; Uzunoglu, S.; Cicin, I. Frequency of RAS Mutations (KRAS, NRAS, HRAS) in Human Solid Cancer. Eurasian J. Med. Oncol. 2017, 1, 1–7. [Google Scholar] [CrossRef]
- Lim, J.P.; Gleeson, P.A. Macropinocytosis: An endocytic pathway for internalising large gulps. Immunol. Cell Biol. 2011, 89, 836–843. [Google Scholar] [CrossRef]
- Nazarenko, I.; Hede, S.M.; He, X.; Hedrén, A.; Thompson, J.; Lindström, M.S.; Nistér, M. PDGF and PDGF receptors in glioma. Ups. J. Med. Sci. 2012, 117, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Quezada, C.; Torres, Á.; Niechi, I.; Uribe, D.; Contreras-Duarte, S.; Toledo, F.; San Martín, R.; Gutiérrez, J.; Sobrevia, L. Role of extracellular vesicles in glioma progression. Mol. Asp. Med. 2018, 60, 38–51. [Google Scholar] [CrossRef]
- Gourlay, J.; Morokoff, A.P.; Luwor, R.B.; Zhu, H.J.; Kaye, A.H.; Stylli, S.S. The emergent role of exosomes in glioma. J. Clin. Neurosci. 2017, 35, 13–23. [Google Scholar] [CrossRef]
- Ha, K.D.; Bidlingmaier, S.M.; Liu, B. Macropinocytosis exploitation by cancers and cancer therapeutics. Front. Physiol. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Giusti, I.; Francesco, M.; Dolo, V. Extracellular Vesicles in Glioblastoma: Role in Biological Processes and in Therapeutic Applications. Curr. Cancer Drug Targets 2017, 17, 221–235. [Google Scholar] [CrossRef]
- Costa Verdera, H.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J. Control. Release 2017, 266, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Maltese, W.A.; Overmeyer, J.H. Methuosis: Nonapoptotic cell death associated with vacuolization of macropinosome and endosome compartments. Am. J. Pathol. 2014, 184, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Shubin, A.V.; Demidyuk, I.V.; Komissarov, A.A.; Rafieva, L.M.; Kostrov, S.V. Cytoplasmic vacuolization in cell death and survival. Oncotarget 2016, 7, 55863–55889. [Google Scholar] [CrossRef]
- Trabbic, C.J.; Dietsch, H.M.; Alexander, E.M.; Nagy, P.I.; Robinson, M.W.; Overmeyer, J.H.; Maltese, W.A.; Erhardt, P.W. Differential induction of cytoplasmic vacuolization and methuosis by novel 2-indolyl-substituted pyridinylpropenones. ACS Med. Chem. Lett. 2014, 5, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Overmeyer, J.H.; Young, A.M.; Erhardt, P.W.; Maltese, W.A. Synthesis and evaluation of indole-based chalcones as inducers of methuosis, a novel type of nonapoptotic cell death. J. Med. Chem. 2012, 55, 1940–1956. [Google Scholar] [CrossRef]
- Rauf, A.; Patel, S.; Imran, M.; Maalik, A.; Arshad, M.U.; Saeed, F.; Mabkhot, Y.N.; Al-Showiman, S.S.; Ahmad, N.; Elsharkawy, E. Honokiol: An anticancer lignan. Biomed. Pharmacother. 2018, 107, 555–562. [Google Scholar] [CrossRef]
- Liou, K.T.; Shen, Y.C.; Chen, C.F.; Tsao, C.M.; Tsai, S.K. The anti-inflammatory effect of honokiol on neutrophils: Mechanisms in the inhibition of reactive oxygen species production. Eur. J. Pharmacol. 2003, 475, 19–27. [Google Scholar] [CrossRef]
- Talarek, S.; Listos, J.; Barreca, D.; Tellone, E.; Sureda, A.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Neuroprotective effects of honokiol: From chemistry to medicine. Biofactors 2017, 43, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Uta, R.; François, C.; Marvin, H.; Philipp, A.; Andrea, P.P.; Henrik, W.; Ralph, L. Anti-inflammatory properties of Honokiol in activated primary microglia and astrocytes. J. Neuroimmunol. 2018, 323, 78–86. [Google Scholar] [CrossRef]
- Lee, Y.J.; Lee, Y.M.; Lee, C.K.; Jung, J.K.; Han, S.B.; Hong, J.T. Therapeutic applications of compounds in the Magnolia family. Pharmacol Ther. 2011, 130, 157–176. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.W.; Chen, J.T.; Hong, C.Y.; Lin, Y.L.; Wang, K.T.; Yao, C.J.; Lai, G.M.; Chen, R.M. Honokiol traverses the blood-brain barrier and induces apoptosis of neuroblastoma cells via an intrinsic bax-mitochondrion-cytochrome c-caspase protease pathway. Neuro Oncol. 2012, 14, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chen, T.L.; Tseng, Y.Y.; Wu, G.J.; Hsieh, M.H.; Lin, Y.W.; Chen, R.M. Honokiol induces autophagic cell death in malignant glioma through reactive oxygen species-mediated regulation of the p53/PI3K/Akt/mTOR signaling pathway. Toxicol. Appl. Pharmacol. 2016, 304, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Shubin, A.V.; Demidyuk, I.V.; Lunina, N.A.; Komissarov, A.A.; Roschina, M.P.; Leonova, O.G.; Kostrov, S.V. Protease 3C of hepatitis A virus induces vacuolization of lysosomal/endosomal organelles and caspase-independent cell death. BMC Cell Biol. 2015, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Recouvreux, M.V.; Commisso, C. Macropinocytosis: A Metabolic Adaptation to nutrient Stress in Cancer. Front. Endocrinol. 2017, 8, 1–7. [Google Scholar] [CrossRef]
- Hanke, J.H.; Gardner, J.P.; Dow, R.L.; Changelian, P.S.; Brissette, W.H.; Weringer, E.J.; Pollok, B.A.; Connelly, P.A. Discovery of a Novel, Potent, and Src Family-selective. J. Biol. Chem. 1996, 271, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Castro-Obregón, S.; Rao, R.V.; Del Rio, G.; Chen, S.F.; Poksay, K.S.; Rabizadeh, S.; Vesce, S.; Zhang, X.K.; Swanson, R.A.; Bredesen, D.E. Alternative, nonapoptotic programmed cell death: Mediation by arrestin 2, ERK2, and Nur77. J. Biol. Chem. 2004, 279, 17543–17553. [Google Scholar] [CrossRef]
- Thomé, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of acridine orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, S.; de Belle, I.; Bredesen, D.E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, X.; Yang, Z.; Zhao, X. Honokiol induces caspase-independent paraptosis via reactive oxygen species production that is accompanied by apoptosis in leukemia cells. Biochem. Biophys. Res. Commun. 2013, 430, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; van den Bent, M.; Hopkins, K.; Tonn, J.C.; Stupp, R.; Falini, A.; Cohen-Jonathan-Moyal, E.; Frappaz, D.; Henriksson, R.; Balana, C.; et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014, 15, 395–403. [Google Scholar] [CrossRef]
- Serventi, J.; Behr, J. Surgery and Evidence-based Treatments in Patients with Newly Diagnosed High-grade Glioma. Semin. Oncol. Nurs. 2018, 34, 443–453. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, Y.P.; Weatherbee, J.L.; Wheelhouse, R.T.; Ross, A.H. Glioblastoma multiforme therapy and mechanisms of resistance. Pharmaceuticals (Basel) 2013, 6, 1475–1506. [Google Scholar] [CrossRef] [PubMed]
- López-Ginés, C.; Navarro, L.; Muñoz-Hidalgo, L.; Buso, E.; Morales, J.M.; Gil-Benso, R.; Gregori-Romero, M.; Megías, J.; Roldán, P.; Segura-Sabater, R.; et al. Association between epidermal growth factor receptor amplification and ADP-ribosylation factor 1 methylation in human glioblastoma. Cell. Oncol. 2017, 40, 389–399. [Google Scholar] [CrossRef]
- Oberbanscheidt, P.; Balkow, S.; Kühnl, J.; Grabbe, S.; Bähler, M. SWAP-70 associates transiently with macropinosomes. Eur. J. Cell Biol. 2007, 86, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Padrick, S.B.; Doolittle, L.K.; Daugherty-Clarke, K.; Corrêa, I.R.; Xu, M.Q.; Goode, B.L.; Rosen, M.K.; Gelles, J. Three-color single molecule imaging shows WASP detachment from Arp2/3 complex triggers actin filament branch formation. Elife 2013, 2, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Egami, Y.; Taguchi, T.; Maekawa, M.; Arai, H.; Araki, N. Small GTPases and phosphoinositides in the regulatory mechanisms of macropinosome formation and maturation. Front. Physiol. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, X.; Chen, C.; Liu, B.; Ren, B.; Wang, L.; Zhao, K.; Yu, S.; Ming, H. Expression of the Arp2/3 complex in human gliomas and its role in the migration and invasion of glioma cells. Oncol. Rep. 2013, 30, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Mariani, L.; Beaudry, C.; McDonough, W.S.; Hoelzinger, D.B.; Demuth, T.; Ross, K.R.; Berens, T.; Coons, S.W.; Watts, G.; Trent, J.M.; et al. Glioma cell motility is associated with reduced transcription of proapoptotic and proliferation genes: A cDNA microarray analysis. J. Neurooncol. 2001, 53, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Gao, Y.; Chen, L.; Li, Y.L.; Jiang, C.L. RAB34 was a progression- and prognosis-associated biomarker in gliomas. Tumor Biol. 2014, 36, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Chen, Q.; Liu, B.; Wang, L.; Zhang, S.; Ji, B. Knockdown of Rab21 inhibits proliferation and induces apoptosis in human glioma cells. Cell. Mol. Biol. Lett. 2017, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, D.; Davidson, A.C.; Hume, P.J.; Makin, L.E.; Koronakis, V. Arf6 coordinates actin assembly through the WAVE complex, a mechanism usurped by Salmonella to invade host cells. Proc. Natl. Acad. Sci. USA 2013, 110, 16880–16885. [Google Scholar] [CrossRef]
- Hu, B.; Shi, B.; Jarzynka, M.J.; Yiin, J.J.; D’Souza-Schorey, C.; Cheng, S.Y. ADP-ribosylation factor 6 regulates glioma cell invasion through the IQ-domain GTPase-activating protein 1-Rac1-mediated pathway. Cancer Res. 2009, 69, 794–801. [Google Scholar] [CrossRef]
- Li, M.; Ng, S.S.M.; Wang, J.; Lai, L.; Leung, S.Y.; Franco, M.; Peng, Y.; He, M.L.; Kung, H.F.; Lin, M.C.M. EFA6A enhances glioma cell invasion through ADP ribosylation factor 6/extracellular signal-regulated kinase signaling. Cancer Res. 2006, 66, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.S.; Aaberg-Jessen, C.; Christensen, K.G.; Kristensen, B. Expression of the lysosomal-associated membrane protein-1 (LAMP-1) in astrocytomas. Int. J. Clin. Exp. Pathol. 2013, 6, 1294–1305. [Google Scholar] [CrossRef]
- Sarafian, V.S.; Koev, I.; Mehterov, N.; Kazakova, M.; Dangalov, K. LAMP-1 gene is overexpressed in high grade glioma. Apmis 2018, 126, 657–662. [Google Scholar] [CrossRef]
- Haga, Y.; Miwa, N.; Jahangeer, S.; Okada, T.; Nakamura, S.I. CtBP1/BARS is an activator of phospholipase D1 necessary for agonist-induced macropinocytosis. EMBO J. 2009, 28, 1197–1207. [Google Scholar] [CrossRef]
- Liberali, P.; Kakkonen, E.; Turacchio, G.; Valente, C.; Spaar, A.; Perinetti, G.; Böckmann, R.A.; Corda, D.; Colanzi, A.; Marjomaki, V.; et al. The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J. 2008, 27, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Di, G. C-Terminal Binding Protein is Involved in Promoting to the Carcinogenesis of Human Glioma. Mol. Neurobiol. 2017, 54, 6121–6132. [Google Scholar] [CrossRef]
- Zhao, C.; Shen, Y.; Tao, X.; Xu, J.; Lu, J.; Liu, C.; Xu, Z.; Tang, Q.; Tao, T.; Zhang, X. Silencing of CtBP1 suppresses the migration in human glioma cells. J. Mol. Histol. 2016, 47, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Yokoyama, T.; Fujiwara, K.; Tari, A.M.; Sawaya, R.; Suki, D.; Hess, K.R.; Aldape, K.D.; Kondo, S.; Kumar, R.; et al. Phosphorylated Pak1 level in the cytoplasm correlates with shorter survival time in patients with glioblastoma. Clin. Cancer Res. 2007, 13, 6603–6609. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, R.J. Driving glioblastoma to drink. Cell 2014, 157, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.; Lee, H.; Song, S.; Kim, N.S.; Im, W.; Kim, M.; Seo, H.; Chung, C.; Chang, J.; Ferrante, R.J.; et al. SCAMP5 Links Endoplasmic Reticulum Stress to the Accumulation of Expanded Polyglutamine Protein Aggregates via Endocytosis Inhibition. J. Biol. Chem. 2009, 284, 11318–11325. [Google Scholar] [CrossRef]
- Gunduz, N.; Ceylan, H.; Guler, M.O.; Tekinay, A.B. Intracellular Accumulation of Gold Nanoparticles Leads to Inhibition of Macropinocytosis to Reduce the Endoplasmic Reticulum Stress. Sci. Rep. 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lin, C.; Lin, Y. Data in Brief Data analyses of honokiol-induced autophagy of human glioma cells in vitro and in vivo. Data Brief 2016, 9, 667–672. [Google Scholar] [CrossRef]
- Sander, P.; Walther, P.; Moepps, B.; Hinz, M.; Mostafa, H.; Schaefer, P.; Pala, A.; Wirtz, R.; Georgieff, M.; Schneider, M. Mitophagy-related cell death mediated by vacquinol-1 and TRPM7 blockade in glioblastoma IV. IntechOpen 2019, 5, 81–93. [Google Scholar] [CrossRef]
- Florey, O.; Overholtzer, M.; Overholtzer, M. Macropinocytosis and autophagy crosstalk in nutrient scavenging. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374. [Google Scholar] [CrossRef]
- Gan, H.K.; Reardon, D.A.; Lassman, A.B.; Merrell, R.; Van Den Bent, M.; Butowski, N.; Lwin, Z.; Wheeler, H.; Fichtel, L.; Scott, A.M.; et al. Safety, Pharmacokinetics and Antitumor Response of Depatuxizumab Mafodotin as Monotherapy or in Combination with Temozolomide in Patients with Glioblastoma. Neuro Oncol. 2018, 20, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Van Den Bent, M.J.; Gan, H.K.; Reardon, D.A.; Kumthekar, P.; Butowski, N.; Lwin, Z.; Mikkelsen, T.; Nabors, L.B.; Kyriakos, P.; et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: Results from an international phase I multicenter trial. Neuro Oncol. 2019, 21, 106–114. [Google Scholar] [CrossRef]
- Wang, X.; Duan, X.; Yang, G.; Zhang, X.; Deng, L.; Zheng, H.; Deng, C.; Wen, J.; Wang, N.; Peng, C.; et al. Honokiol Crosses BBB and BCSFB, and Inhibits Brain Tumor Growth in Rat 9L Intracerebral Gliosarcoma Model and Human U251 Xenograft Glioma Model. PLoS ONE. 2011, 6, e18490. [Google Scholar] [CrossRef]
- Li, Z.; Mbah, N.E.; Overmeyer, J.H.; Sarver, J.G.; George, S.; Trabbic, C.J.; Erhardt, P.W.; Maltese, W.A. The JNK signaling pathway plays a key role in methuosis (non-apoptotic cell death) induced by MOMIPP in glioblastoma. BMC Cancer 2019, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Ahlstedt, J.; Förnvik, K.; Zolfaghari, S.; Kwak, D.; Lars, G.J.; Ernfors, P.; Salford, L.G.; Redebrandt, H.N. Evaluating vacquinol-1 in rats carrying glioblastoma models RG2 and NS1. Oncotarget 2018, 9, 8391–8399. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.C.; Tai, Y.T.; Mohanraj, M.; Liu, S.H.; Yang, S.T.; Chen, R.M. Honokiol enhances temozolomide-induced apoptotic insults to malignant glioma cells via an intrinsic mitochondrion-dependent pathway. Phytomedicine 2018, 49, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Xue, W.; Schachner, M.; Zhao, W. Honokiol Eliminates Glioma/Glioblastoma Stem Cell-Like Cells via JAK-STAT3 Signaling and Inhibits Factor Receptor. Cancer 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ballesta, A.; Zhou, Q.; Zhang, X.; Lv, H.; Gallo, J. Multiscale Design of Cell-Type–Specific Pharmacokinetic/ Pharmacodynamic Models for Personalized Medicine: Application to Temozolomide in Brain Tumors. CPT Pharmacomet. Syst. Pharmacol. 2014, 4, 1–11. [Google Scholar] [CrossRef]
- Sun, L.; Hui, A.; Su, Q.; Vortmeyer, A.; Kotliarov, Y.; Pastorino, S.; Passaniti, A.; Menon, J.; Walling, J.; Bailey, R.; et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 2006, 9, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Yamamoto, H.; Suetsugu, S.; Miki, H.; Takenawa, T.; Endo, T. Small GTPase Rah/Rab34 is associated with membrane ruffles and macropinosomes and promotes macropinosome formation. J. Biol. Chem. 2003, 278, 4063–4071. [Google Scholar] [CrossRef] [PubMed]
- Bhanot, H.; Young, A.M.; Overmeyer, J.H.; Maltese, W.A. Induction of Nonapoptotic Cell Death by Activated Ras Requires Inverse Regulation of Rac1 and Arf6. Mol. Cancer Res. 2010, 8, 1358–1374. [Google Scholar] [CrossRef]
- Fujii, M.; Kawai, K.; Egami, Y.; Araki, N. Dissecting the roles of Rac1 activation and deactivation in macropinocytosis using microscopic photo-manipulation. Sci. Rep. 2013, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Koronakis, V.; Hume, P.J.; Humphreys, D.; Liu, T.; Horning, O.; Jensen, O.N.; McGhie, E.J. WAVE regulatory complex activation by cooperating GTPases Arf and Rac1. Proc. Natl. Acad. Sci. USA 2011, 108, 14449–14454. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, W.D.; Yoshida, S.; Straight, S.W.; Swanson, J.A. Coordination of the Rab5 Cycle on Macropinosomes. Traffic 2011, 12, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.L.; Cao, C.; Pylypenko, O.; Rak, A.; Wandinger-Ness, A. Rab GTPases at a glance. J. Cell Sci. 2008, 121, 246. [Google Scholar] [CrossRef]
- Egami, Y.; Araki, N. Rab20 regulates phagosome maturation in RAW264 macrophages during Fc gamma receptor-mediated phagocytosis. PLoS ONE 2012, 7, e35663. [Google Scholar] [CrossRef]
- Teasdale, R.D.; Loci, D.; Houghton, F.; Karlsson, L.; Gleeson, P.A. A large family of endosome-localized proteins related to sorting nexin 1. Biochem. J. 2001, 358, 7–16. [Google Scholar] [CrossRef]
- Merino-Trigo, A.; Kerr, M.; Houghton, F.; Lindberg, A.; Mitchell, C.; Teasdale, R.; Gleeson, P. Sorting nexin 5 is localized to a subdomain of the early endosomes and is recruited to the plasma membrane following EGF stimulation. J. Cell Sci. 2004, 117, 6413–6424. [Google Scholar] [CrossRef]
- Kerr, M.C.; Lindsay, M.; Luetterforst, R.; Hamilton, N.; Simpson, F.; Parton, R.; Gleeson, P.; Teasdale, R. Visualisation of macropinosome maturation by the recruitment of sorting nexins. J. Cell Sci. 2006, 119, 3967–3980. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colin, M.; Delporte, C.; Janky, R.; Lechon, A.-S.; Renard, G.; Van Antwerpen, P.; Maltese, W.A.; Mathieu, V. Dysregulation of Macropinocytosis Processes in Glioblastomas May Be Exploited to Increase Intracellular Anti-Cancer Drug Levels: The Example of Temozolomide. Cancers 2019, 11, 411. https://doi.org/10.3390/cancers11030411
Colin M, Delporte C, Janky R, Lechon A-S, Renard G, Van Antwerpen P, Maltese WA, Mathieu V. Dysregulation of Macropinocytosis Processes in Glioblastomas May Be Exploited to Increase Intracellular Anti-Cancer Drug Levels: The Example of Temozolomide. Cancers. 2019; 11(3):411. https://doi.org/10.3390/cancers11030411
Chicago/Turabian StyleColin, Margaux, Cédric Delporte, Rekin’s Janky, Anne-Sophie Lechon, Gwendoline Renard, Pierre Van Antwerpen, William A. Maltese, and Véronique Mathieu. 2019. "Dysregulation of Macropinocytosis Processes in Glioblastomas May Be Exploited to Increase Intracellular Anti-Cancer Drug Levels: The Example of Temozolomide" Cancers 11, no. 3: 411. https://doi.org/10.3390/cancers11030411