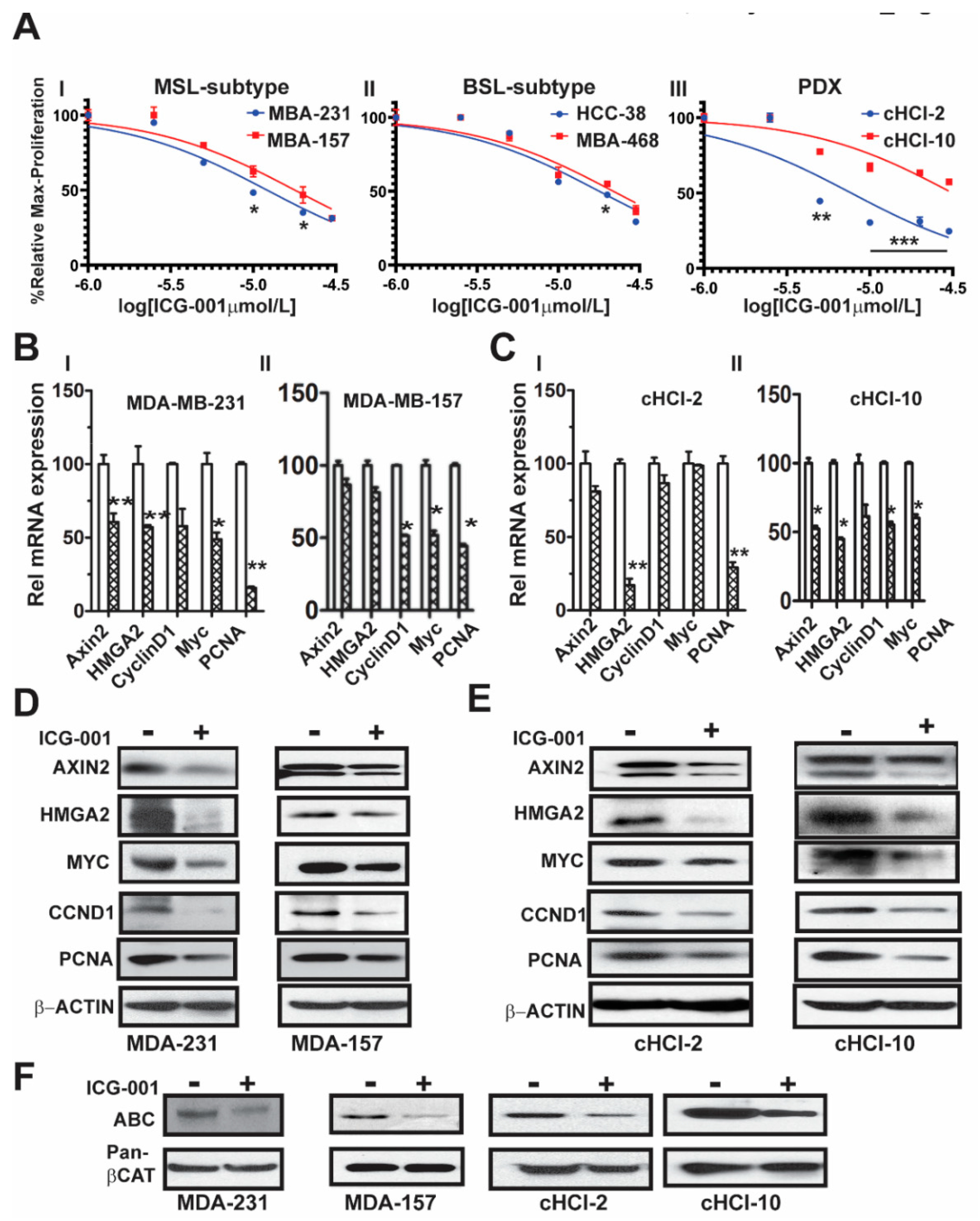
2.1. The WNT Inhibitor ICG-001 Inhibits the Growth and β-Catenin Gene Transcription of TNBC Cells
We previously demonstrated that WNT10B has epistatic activity on HMGA2 in MDA-MB-231 cells, regulating WNT10B-mediated proliferation [
9]. Moreover, we showed that ICG-001 targets an auto-regulatory-loop, composed of HMGA2 and EZH2, that is necessary for maintenance of the WNT nuclear components β-catenin/TCF4/LEF-1 to induce gene transcription of WNT10B direct target genes [
10]. To determine the effects of ICG-001 on proliferation of a subset of TNBC cell lines, we directly compared cell lines from two different TNBC subtypes, mesenchymal stem like (MSL: MDA-MB-231 and MDA-MB-157) and basal-like 1 (BL1: HCC-38 and MDA-MB-468). We also included cells derived from two TNBC PDX tumor models, one chemo-naïve (cHCI-2, never exposed to therapy) and one highly resistant to doxorubicin (cHCI-10)
Figure 1A. Utilizing WST-1 proliferation assays, we determined the IC
50 dose for ICG-001 following 48 h of exposure to increasing dosages ranging from 0.02 µM to 30 µM (0.02, 0.04, 0.2, 1, 5, 10, 20 and 30 µM). The ICG-001 effects are specific for TNBC, as exposure of ICG-001 was nontoxic to normal human mammary epithelial cells (HUMEC), and human epithelial breast MCF10A cells, as well as ER+ MCF7 and HER2+ SKBr3 cell lines (
Figure S1A). The cHCI-2 chemo-naïve cells were the most sensitive to ICG-001, whereas the chemo-resistant cHCI-10 cells were the most resistant. The mean IC
50 levels for all cell lines are shown in
Figure S1B; all downstream experiments were conducted with the IC
50s shown in
Figure S1B.
Next, to determine the effects of ICG-001 on known WNT10B/β-catenin direct target genes (
AXIN2 and
HMGA2) [
9,
10] and proliferation-associated genes (
CCND1, MYC, and
PCNA), we treated the cells at their respective IC
50 concentrations for 48 h as follows: MDA-MB-231 (10 μM) and MDA-MB-157 (20 μM,
Figure 1BI,II) or cHCI-2 (6 μM) and cHCI-10 (35 μM,
Figure 1CI,II) cells and then conducted qPCR analysis (
Table S1). There was a significant downregulation of
HMGA2 mRNA in MDA-MB-231, cHCI-2 and cHCI-10 cells (*
p < 0.05 or **
p < 0.01). In contrast, the MDA-MB-157 cells did not show a significant change. Interestingly, MDA-MB-231 cells and cHCI-10 cells had a similar significant downregulation of mRNA expression for
CCND1, MYC and
PCNA (*
p = 0.05 to **
p = 0.01) and these results were confirmed by immunoblotting (
Figure 1D,E). In MDA-MB-157 cells, the mRNA expression was reduced significantly only for
CCND1,
MYC and
PCNA (
* p = 0.01) and this was confirmed by immunoblotting.
The above results suggest that ICG-001 repressed WNT direct target genes by the disruption of transcriptionally active β-catenin, also known as ABC (i.e., lacks phosphorylation at amino acids Ser33/Ser37/Thr41) that is co-localized in the nucleus [
12]. To test for that possibility, we performed immunoblotting for ABC in MDA-MB-231, MDA-MB-157, cHCI-2 and cHCI-10 cells at the appropriate IC
50 dose for 48 h (
Figure 1F). In response to ICG-001, ABC protein expression is decreased in the TNBC cell lines. Pan-β-catenin and β-actin served as controls. Immunoblots were quantified in a set of biological triplicates that demonstrated statistically significant changes of the immunoblots when treated with ICG-001 relative to the controls for each cell line tested (
Figure S1Ci,ii). Taken together, the data suggest that ICG-001 decreases proliferation and reduces the expression of WNT10B/β-catenin direct target genes in a variety of TNBC cell lines.
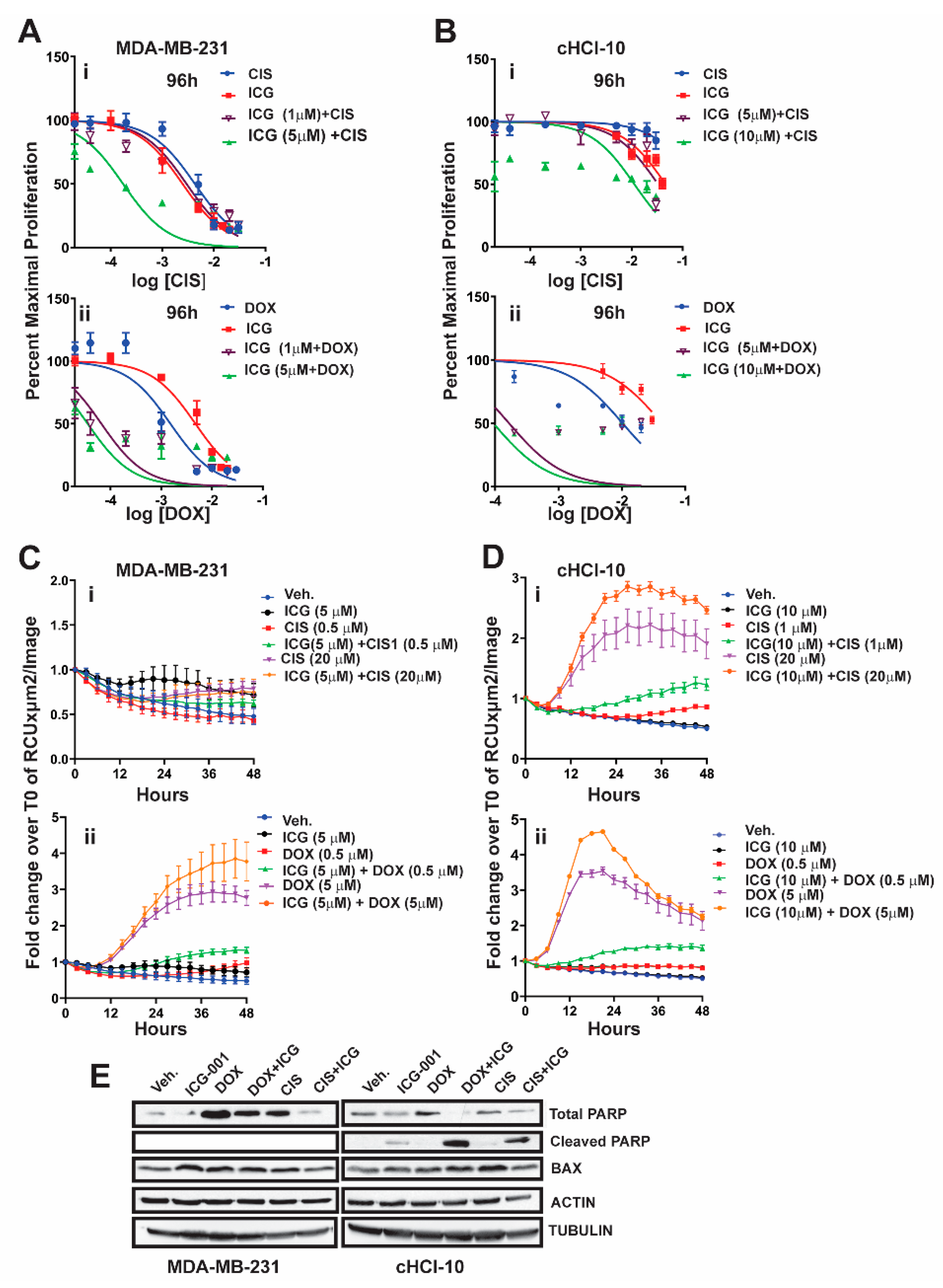
2.2. The WNT Inhibitor ICG-001 Preferentially Synergizes with Doxorubicin, But Not Cisplatin, in Highly Chemo-Resistant TNBC Cells
It is known that β-catenin contributes to resistance to doxorubicin and to cisplatin in MDA-MB-231 cells, as the silencing of β-catenin expression restores drug sensitivity [
13]. We have shown that, in an TNBC cell line (cHCI-10), ICG-001 is capable of sensitizing cells to doxorubicin and that this effect was synergistic by isobologram and combination index analysis [
10]. However, the ability of ICG-001 to sensitize these cells to another FDA-approved frontline chemotherapy drug against TNBC, such as cisplatin (CIS), is unknown.
To test for synergy of ICG-001 with cisplatin, we next used sub-IC
50 concentrations of ICG-001, either at 1 µM or 5 µM, for MDA-MB-231 cells, or at 5 µM and 10 µM for cHCI-10 cells in combination with various CIS dosages ranging from 0.02 μM to 20 μM (0.02, 0.04, 0.2, 1, 5, 10, and 20 μM) (
Figure 2). WST-1 proliferation assays were performed every 24 h up to 96 h (
Figure 2Ai or
Figure 2Bi) and 48 h is shown in
Figure S2Ai. In MDA-MB-231 cells, ICG-001 (5 μM) was capable of reaching the IC
50 threshold with as little as 0.04 μM of CIS by 96 h. As a monotherapy, CIS required 125-fold (5 μM) more drug to reach the IC
50 threshold by 96 h. In contrast, ICG-001 was not able to reach the IC
50 threshold in combination with CIS in the cHCI-10 cells until the ranges reached 20 μM by 96 h. Similar effects were observed in the cHCI-10 cells at 48 h (
Figure S2Bi). The patient from which the HCI-10 model was derived was not treated with a platinum agent, but died after treatment with cyclophosphamide, anthracyclines and taxanes [
14]. Of note, there were no effects on proliferation with the drug combination at 24 h in either the MDA-MB-231 or cHCI-10 cell lines. The above data suggest that ICG-001 was more effective at sensitizing CIS-based therapy in the MDA-MB-231 cells than in the PDX-derived cells. We determined IC
30, IC
50 and IC
70 values and plotted isobole curves to determine the combination index, but we found no additive or synergistic effects between cisplatin with ICG-001 [
10,
15] as the combination indexes were greater than 1.
Next, we repeated the above experiment with the same sub-IC
50 concentrations of ICG-001 with increasing DOX dosages ranging from 0.02 to 20 μM (0.02, 0.04, 0.2, 1, 2, 10 and 20 μM) for both MDA-MB-231 and cHCI-10 cells (
Figure 2A–D). In the MDA-MB-231 cells, the sub-IC
50 concentrations of ICG-001 (1 μM and/or 5 μM) were capable of sensitizing cells to DOX therapy at 0.04 μM by 96 h (
Figure 2Aii). In contrast, the DOX alone treatment required 25-fold higher concentrations (1 μM) to reach this threshold. There were no effects on proliferation at 24 h. The 48-h time point is shown for the MDA-MB-231 cells in
Figure S2Ai, demonstrating similar trends as the 96-h analysis.
The cHCI-10 cells were found to be more sensitive to the combinatorial therapies of ICG-001 plus DOX reaching the sub-IC
50 threshold (~48–49%) (
Figure 2). Importantly, at 96 h, ICG-001 was able to sensitize DOX with the three lowest concentrations of DOX (0.02, 0.04 and 0.2 μM;
Figure 2Aii,Bii). The DOX alone treatment required a range of 50–500-fold more drug, depending on the dosage comparisons, than the combination therapies to reach sub-IC
50 thresholds in cHCI-10 cells at the same time. Similar trends were observed at the 48-h time point (
Figure S2Ai,Bii). Taken together, these results suggest that WNT-therapy can sensitize the TNBC PDX cells to doxorubicin, but not to cisplatin. We have previously published the IC
30, IC
50 and IC
70 values and plotted isobole curves to determine the combination index; the combination indexes for doxorubicin and ICG-001 were less than 1, confirming synergistic effects [
10,
15].
Next, we conducted assays using the IncuCyte
® Cytotox Green Reagent that quantifies cell death in real time. We used sub-IC
50 concentrations of ICG-001, either at 5 µM, for MDA-MB-231 cells, or at 10 µM for cHCI-10 cells in combination with two CIS dosages at 0.5 µM or 20 µM in the MDA-MB-231 (
Figure 2Ci) and 1 µM or 20 µM for the cHCI-10 cells for 48 h (
Figure 2Di). Similarly, we used two DOX dosages 0.5 µM or 5 µM in the MDA-MB-231 cells (
Figure 2Cii) and in the cHCI-10 cells (
Figure 2Dii). The results show that the amplitude of the cytotoxicity effect of the combinatorial treatment is highest when cells are treated with DOX plus ICG-001 in both cell lines. In contrast, CIS plus ICG-001 was not effective in the MDA-MB-231 cells, but was in the cHCI-10 cells. It is known that CIS can elicit chemoresistance through hyper activity of PARP-1 in non-small cell lung carcinomas [
16] and that PARP-1 regulates EMT through SNAIL expression in doxorubicin-resistant MDA-MB-231 cells [
17]. We posited that PARP expression could account for the cell death difference between the two models above; thus, we immunoblotted for total PARP-1 and cleaved PARP-1 (
Figure 2F). Results indicate that both DOX and CIS induce PARP-1 protein expression in MDA-MB-231 cells as expected, consistent with the literature. In contrast, this is not observed in cHCI-10 cells. More importantly, ICG-001 therapy sensitizes with both CIS and DOX to generate the cleaved PARP-1 product, but not in the MDA-MB-231 cells. BAX serves as a cell death marker control and both ACTIN and TUBULIN serve as loading controls. These data support the argument that DOX plus ICG-001 synergize to inhibit proliferation and to activate cell death by inhibition of PARP-1 activity.
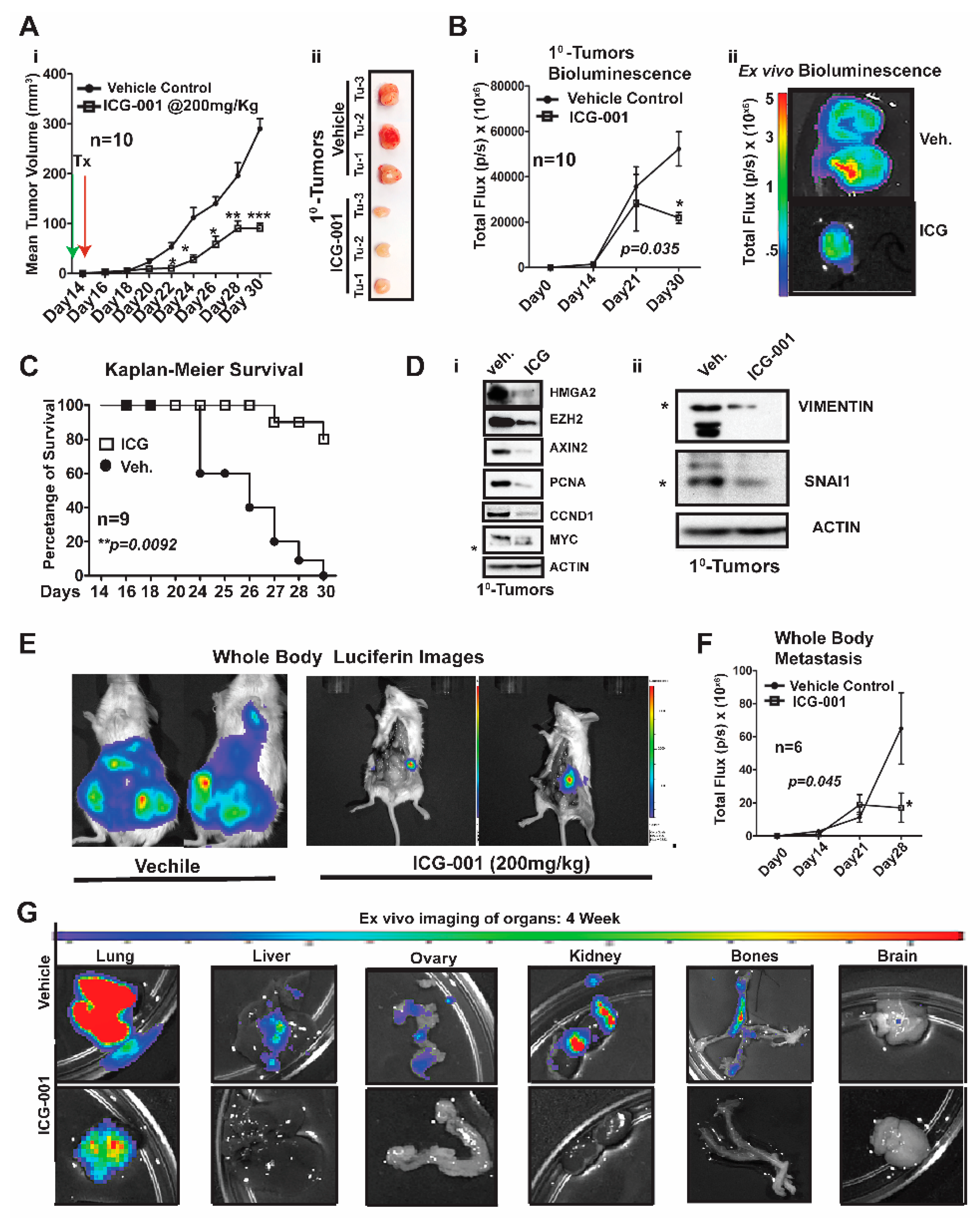
2.3. WNT Inhibition Interferes with Simultaneous Multi-Organ Metastases in Vivo in MDA-MB-231 Cells
Over a decade ago, the Massague lab characterized MDA-MB-231 metastatic models generated by intracardiac (IC) xenografting, demonstrating that these same cells would show enhanced metastasis to the lung and bone after simultaneous repeated IC injections [
18]. More importantly, they demonstrated that WNT signaling was critical for maintenance of the aforementioned metastatic enrichment models [
19]. We questioned if we could: (1) Improve metastatic modeling by surgically transplanting MDA-MB-231 cells directly into the mammary fat of immunocompromised NOD/SCID Gamma (NSG) mice. (2) Generate de novo simultaneous multi-organ metastases, including lymph node and bone metastases, and (3) address one of the overarching challenges of MBC and/or metTNBC, which is the inability to treat widespread metastases, including to visceral organs (liver and lungs).
We first generated MDA-MB-231-Luc cells from parental MDA-MB-231 (stably transfected with a lentiviral luciferase vector) and bilaterally surgically transplanted ~1.25 × 10
6 cells into the mammary fat pad of NSG mice (
Figure 3). One week after surgery, we began treatment with ICG-001 via intraperitoneal injections every other day for two weeks (200 mg/kg, IP; n = 10 mice/group). This dosage of ICG-001 is the human equivalent dosage (HED) of 600 mg/m
2, which is currently used in a phase II clinic trials (
https://clinicaltrials.gov/), ICG-001/PRI-724;
Figure 3Ai). We determined that relative to vehicle (Veh) treatment, ICG-001 significantly decreased tumor volume (mm
3) as early as day 22 post-transplantation (***
p = 0.001), and a reduced tumor volume is maintained through the study endpoint (day 30 post-transplantation). At tumor harvest, we obtained ex vivo images of control and ICG-001-treated tumors, showing reduced overall size and angiogenesis in the ICG-001-treated tumors (
Figure 3Aii; n = 3). We also longitudinally compared tumor bioluminescence flux for all the primary tumors throughout the study, since bioluminescence activity requires ATP from live cells, confirming that tumors treated with ICG-001 had reduced viability (
Figure 3Bi;
p = 0.048). These observations were verified by ex vivo bioluminescence images of mice treated with ICG-001 therapy relative to the vehicle controls. Additionally, the average endpoint tumor wet weight was significantly reduced in the ICG-001 cohort, whereas there were no significant changes in the mean animal body weights in response to therapy (
Figure S3Ai,ii).
Untreated mice were removed from the study due to either primary tumor burden, metastatic burden and/or or deterioration of health status (the mouse was then scored as dead). Based on these criteria for survival, the Kaplan–Meier curves show that ICG-001 significantly increased survival rates (80%) (
Figure 3C;
p = 0.0092). Mechanistically, in these tumors, WNT10B/β-CATENIN direct target genes [
9,
10,
20] (AXIN2, HMGA2, CCND1, PCNA, and MYC;
Figure 3Di) and EMT markers (Vimentin and SNAIL;
Figure 3Dii) showed decreased protein expression after ICG-001 therapy. Quantification of the immunoblots by ImageJ, from biological triplicate tumors, revealed that ICG-001 reduced protein levels of the aforementioned genes (
Figure S3B). Overall, these in vivo results are congruent with the in vitro results previously shown in
Figure 1 and
Figure S1Ci–ii.
To determine if ICG-001 could inhibit de novo simultaneous multi-organ metastases (lymph node, bone, and brain metastases) observed in our luciferase labeled MDA-MB-231 model, the frequency of metastasis to various organs was compared by conducting ex vivo bioluminescence analysis (
Figure S3C). In untreated cohorts, the lymph nodes, lungs and bones had a metastasis rate of 100% and other organs varied from 80% to 20% frequency. As representative images, we show two whole-body bioluminescence images from two mice each from the vehicle and treated groups (
Figure 3E). ICG-001 therapy significantly decreased whole-body bioluminescence (
Figure 3F) and the number of mice with metastasis (
Figure S3D). Moreover, bioluminescence images of ex vivo organs are shown pre- and post-therapy (
Figure 3G and
Figure S3E). ICG-001 significantly repressed lung, liver, ovary and kidney metastasis (**
p = 0.001;
Figure S3F).
Taken together, these results show that our model generates de novo simultaneous lymph node, visceral organ, bone and brain metastases following orthotopic transplantation of the labeled MDA-MB-231 cells into the mammary fat pad of NSG mice. More importantly, WNT inhibition mediated by ICG-001 monotherapy diminished or blocked metastasis to multiple organs, including the visceral organs typical of TNBC patients (lung and liver).
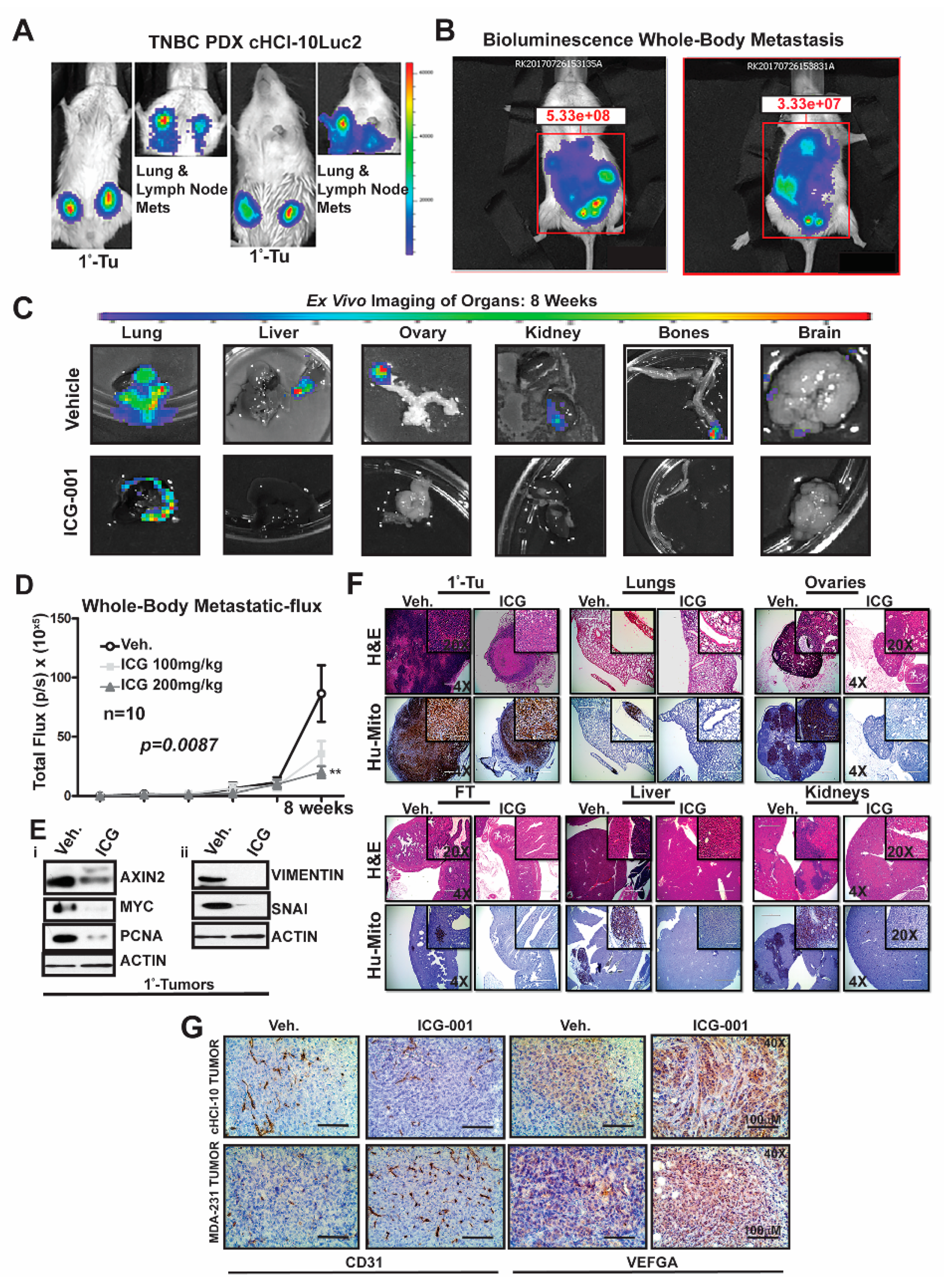
2.4. WNT Inhibition Interferes with De Novo Multi-Organ Metastases in a Chemo-Resistant TNBC PDX Model
To determine the frequency of de novo simultaneous multi-organ metastases from our luciferase-labeled TNBC chemo-resistant PDX tumor model (HCI-10Luc2) [
10], we surgically implanted tumor fragments (~2 mm from a tumor-bearing donor female) into each inguinal mammary gland of 25 NSG mice. Eight weeks after transplantation, bioluminescence imaging was initiated to monitor primary tumor growth with accompanying metastases, which first developed in the axillary lymph nodes (LN) and the lungs, as previously reported
Figure 4A [
10]. We show two representative mice that exhibited metastases affecting multiple organs (
Figure 4B). Ex vivo bioluminescence confirmed metastases in the lung, liver, ovaries, kidneys, bones and brain of these mice (
Figure 4C). The frequency for de novo simultaneous metastases is as follows: lung and LN (100%), ovaries (40%), liver, spleen and kidneys (20%), bone (15%) and brain at 5% (
Figure S4A). More importantly, following ICG-001 therapy at 200 mg/kg (IP, every other day for two weeks), the metastatic flux measured ex vivo was reduced in most of the organs. ICG-001 also reduced primary tumor growth in a statistically significant manner, as shown by: (1) whole tumor images (Bi) (2) bioluminescence imaging of tumors (Bii) and (3) H&E staining (Biii) (
Figure S4B). Tumor growth reduction was observed whether monitored by calipers (Ci), by luciferase flux (Cii) or by tumor wet weight and two images of mice with measurable luciferase flux are shown (
Figure S4C;
* p < 0.05, **
p = 0.001 and
Figure S4D).
Next, we compared whole-body metastases in vehicle control mice vs. ICG-001-treated mice dosed at either 100 mg/kg or 200 mg/kg (n = 10/cohort). The higher dosage resulted in statistically significant repression of luciferase flux units (
Figure 4D; **
p = 0.0087). Mechanistically, the reduction in metastases is associated with loss of protein expression of both WNT10B/β-catenin direct target genes (AXIN2, MYC and PCNA;
Figure 4Ei) and EMT markers (Vimentin and SNAI;
Figure 4Eii) in the primary tumors. Quantification of immunoblots was conducted with ImageJ as previously shown (
Figure S4E). H&E staining of primary HCI-10Luc2 tumors and the simultaneously arising multi-organ metastases in the lungs, ovaries and fallopian tube (FT), liver and kidneys are shown for both the vehicle controls and treated groups (
Figure 4F; 200 mg/kg). Loss of tumor masses in the various organs was confirmed by a reduced immunohistochemistry (IHC) signal for an anti-human mitochondrion (Hu-Mito) antibody, in the aforementioned organs.
Next, we posited that ICG-001 targets non-tumor cell mechanisms are also required for tumor growth and metastasis, such as in endothelial cells. To this end, we conducted immunohistochemistry (IHC) for CD31, a marker for endothelial cells (
Figure 4G). The results showed that CD31 expression was relatively unchanged in the cHCI-10 model. In contrast, CD31 expression was upregulated in the MDA-MB-231 model in response to WNT monotherapy. Based on the above results, we used VEGFA as a marker for angiogenesis to determine if similar results would be observed, as those of CD31. VEGFA protein expression was upregulated in both models after WNT monotherapy. These results suggest that ICG-001 treatment is promoting angiogenesis during tumor outgrowth, perhaps normalizing the tumor vasculature, leading to better tumor perfusion with systemic targeting agents [
21].
The above results present strong evidence that in a highly chemo-resistant TNBC PDX model, transplantation to the mammary fat pad of NSG mice can elicit de novo simultaneous multi-organ metastases. Moreover, for the first time, we demonstrate that ICG-001 monotherapy has the capacity to prevent de novo simultaneous multi-organ metastases arising in this doxorubicin-resistant TNBC PDX model. These data suggest that ICG-001 may be appropriate as a second-line therapy in TNBC patients who progress on anthracyclines.
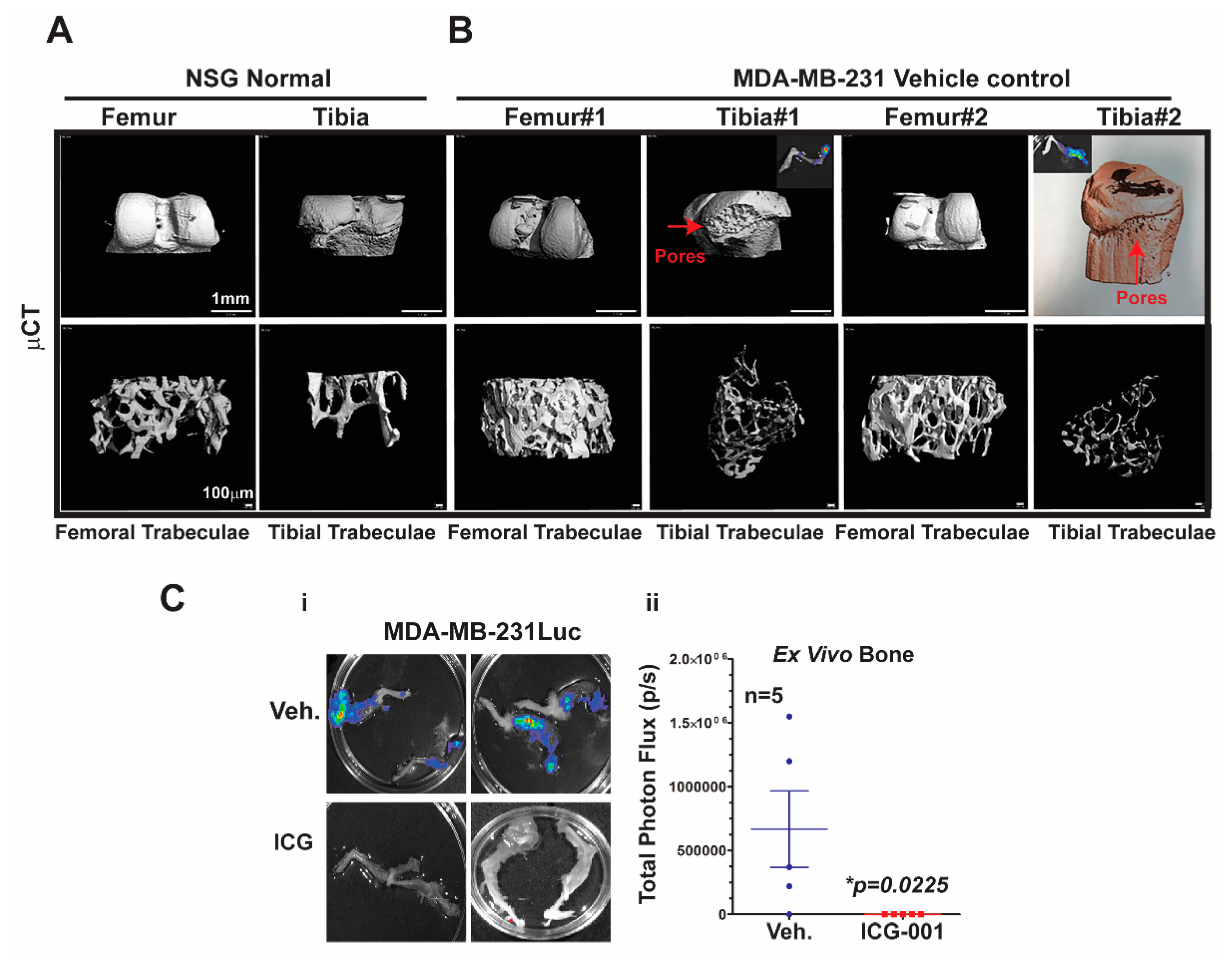
2.5. Micro-Computed Tomography (μCT) Analysis of Bone Metastasis Reveals Differential Osteoclastic Properties in the MDA-MB-231 and TNBC PDX Tumors
Bone metastasis is the most common site of metastasis in women diagnosed with breast cancer, although it is less common in TNBC compared to ER+ breast cancer [
4]. As determined by ex vivo bioluminescence imaging, MDA-MB-231Luc cells surgically transplanted into the mammary fat pad of NSG mice metastasized with 100% frequency to at least one leg bone, and >90% of the time to both legs
Figure 5C.
To characterize the effects of metastasis on bone structure, we conducted micro-computed tomography (μCT) analysis of the bones. First, we demonstrated the baseline μCT for the NSG control female mice
Figure 5A. Both femoral and tibial trabeculae are presented. The μCT of bone metastases derived from MDA-MB-231Luc cells revealed osteolytic properties in the tibial trabeculae, but not the femoral trabeculae (
Figure 5B; n = 2 mice). The red arrows highlight the pores that are generated in the bone by the bone metastasis; the insert shows the corresponding ex vivo bioluminescence from those same bones. Moreover, one of the tibial trabeculae μCT images from the MDA-MB-231 model was pseudo-colored to further highlight the bone resorption. More importantly, ICG-001 therapy prevented bilateral bone metastases arising from the MDA-MB-231Luc cells (
Figure 5Ci,ii; *
p = 0.0225; n = 5 mice). Taken together, these results suggest that bone metastases arising from MDA-MB-231Luc cells respond to WNT monotherapy, as previously observed for lymph node and visceral organ metastases.
2.6. Combination of the WNT Inhibitor ICG-001 and Doxorubicin Blocks Simultaneous Multi-Organ and Bone Metastases in A Chemo-Resistant PDX Model
We next questioned if ICG-001 therapy used in combination with doxorubicin would simultaneously prevent multi-organ metastases and bone metastases originating from the chemo-resistant TNBC PDX tumors. To more efficiently model metastasis, we hypothesized that tail vein injections may increase the experimental-induced metastatic frequency to the bone and other organs (e.g., liver and ovaries) relative to when cells are orthotopically placed into the mammary gland. Therefore, in a pilot experiment, we conducted tail vein injections to experimentally induce metastasis using freshly disassociated HCI-10 Luc tumor cells from multiple pooled HCI-10Luc2 PDX mammary tumors. Cells were digested into small organoids, briefly cultured (2 passages) in M87 media in ultralow adhesion vessels, and then trypsinized and DNase I-treated to obtain a single cell suspension to inject via the tail vein of NSG female mice (1 × 10
6 cells/mouse) (
Figure S5 n = 10 mice). The mice were tracked in vivo for bioluminescence signal longitudinally for ~6–8 weeks and then all organs were harvested and bio-imaged ex vivo. Using this approach, we observed by ex vivo bioluminescence imaging that ~80% of the bones had detectable metastasis. Additionally, we observed an increased frequency of metastasis to the liver and ovaries, amongst other organs and, as expected, 100% of lungs had detectable metastasis (
Figure S5A). These data provide evidence that tail vein injections with cHCI-10Luc cells increase bone metastasis frequency.
Next, we repeated the cHCI-10Luc2 tumor cell isolation process and tail vein injection for the purposes of testing combinatorial therapy with ICG-001 and DOX in vivo (
Figure 6). To limit the potential toxicity of combinatorial therapy, beginning one day after tail vein injection, we treated mice with 50 mg/kg ICG-001 (IP, every other day) and with 1.4 mg/kg of DOX (IP, once every two weeks) for up to three cycles of combination therapy. At these dosages, we have previously shown that the photon flux derived from lung metastases was repressed [
10]. We observed a decrease in total body bioluminescence photon flux in the ICG-001 + DOX cohort relative to the DOX-alone group (
Figure S6B,C; n = 5 per group). Panels of the bioluminescence images are shown from mice in the vehicle, DOX-alone and combination therapy groups.
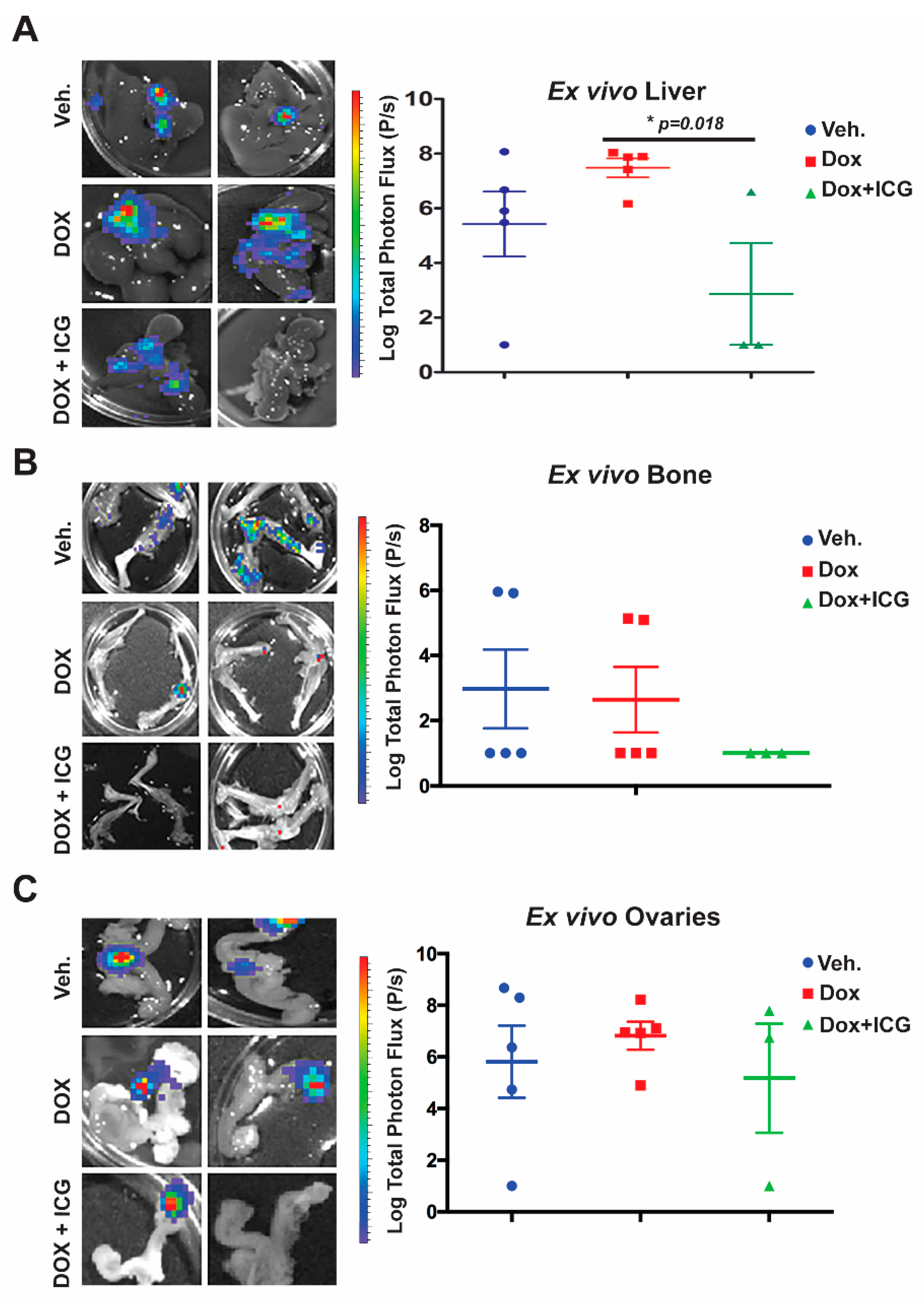
Next, we conducted ex vivo bioluminescence of the liver, bone and ovaries to quantitate photon flux at the study endpoint (
Figure 6A–C). As shown in the representative images from each organ for the above three cohorts, with DOX alone, there was no significant difference in the liver, bone or ovary’s metastatic signal (
Figure 6A–C) relative to the vehicle control. Moreover, liver metastasis for the DOX-alone cohort had statistically higher flux-units versus vehicle-treated mice (
Figure 6A.) In contrast, the DOX + ICG-001 combinatorial therapy significantly repressed liver flux compared to DOX-alone, but combination therapy did not significantly repress metastatic flux in the ovaries or bone relative to DOX alone (
Figure 6B,C). These results suggest that liver, responds to the combinatorial therapy, as was the case with the lungs [
10]. Similarly, the kidneys showed repression of metastases with the combinatorial therapies with lungs serving as the positive control (
Figure S6D,E).
Taken together, these results demonstrate that the addition of a WNT inhibitor to anthracycline therapy can inhibit simultaneous multi-organ metastases, including bone metastases, in a chemo-resistant TNBC PDX model.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





