Targeted-Gene Sequencing to Catch Triple Negative Breast Cancer Heterogeneity before and after Neoadjuvant Chemotherapy

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
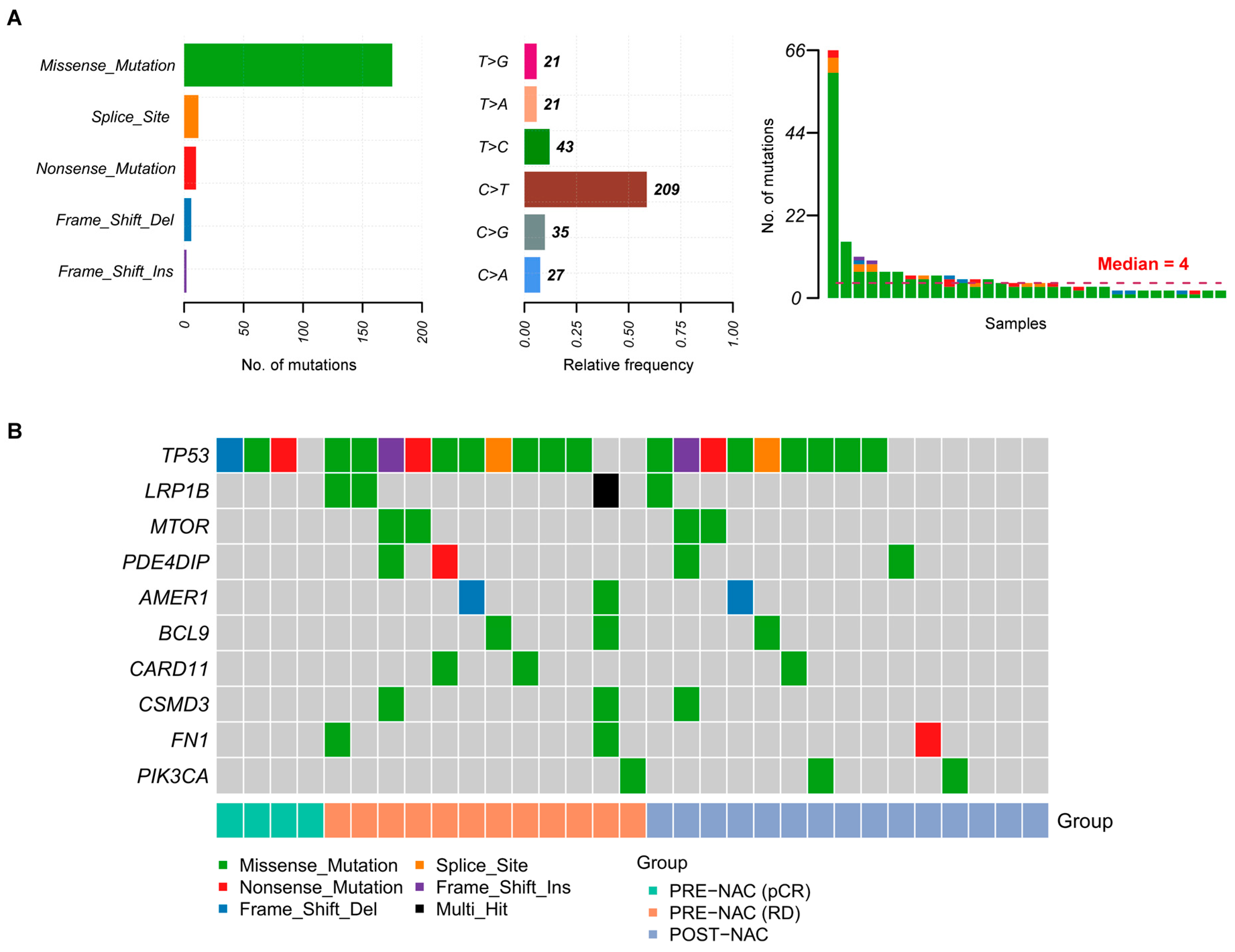
2.2. Somatic Mutations in TNBC Samples
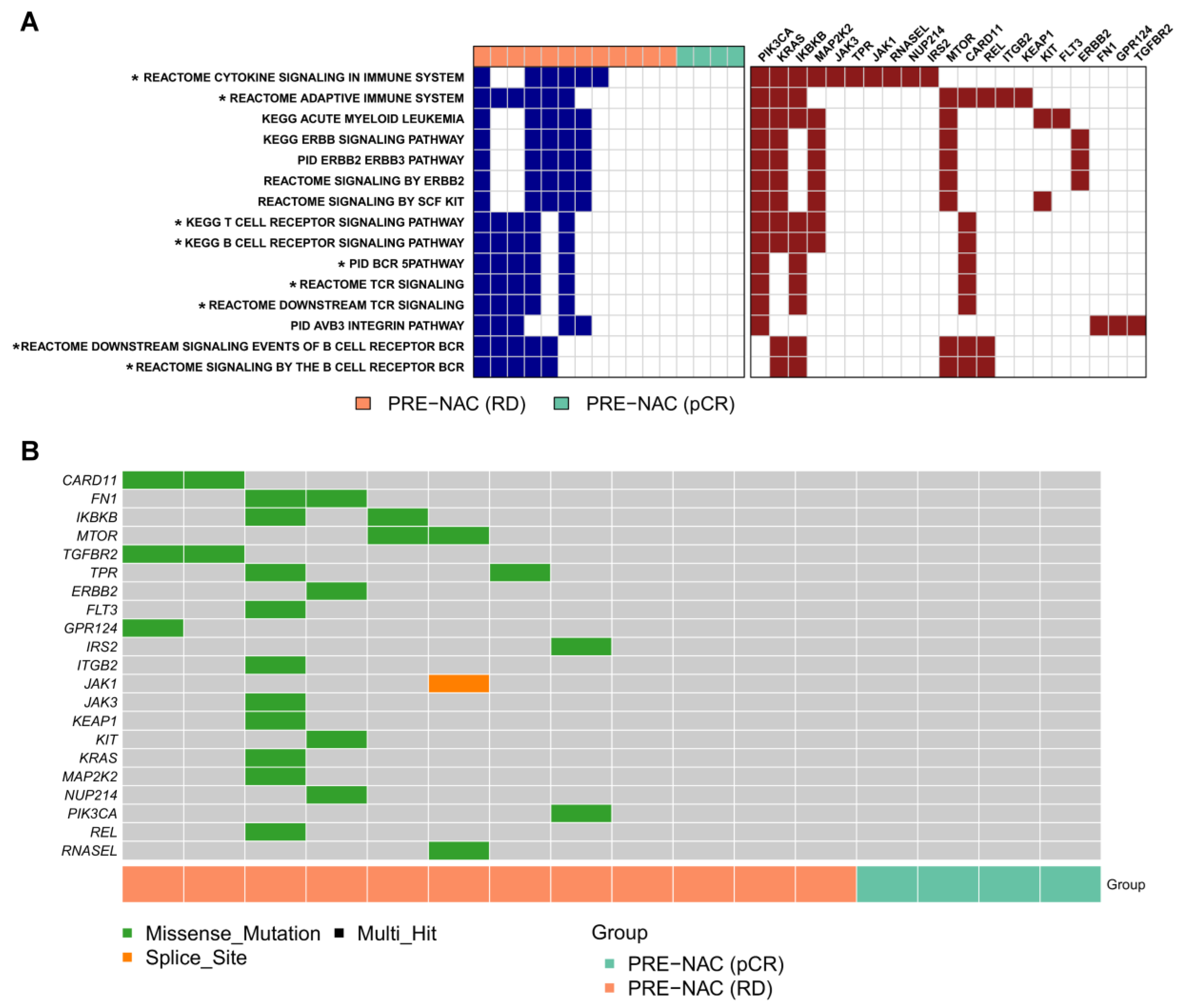
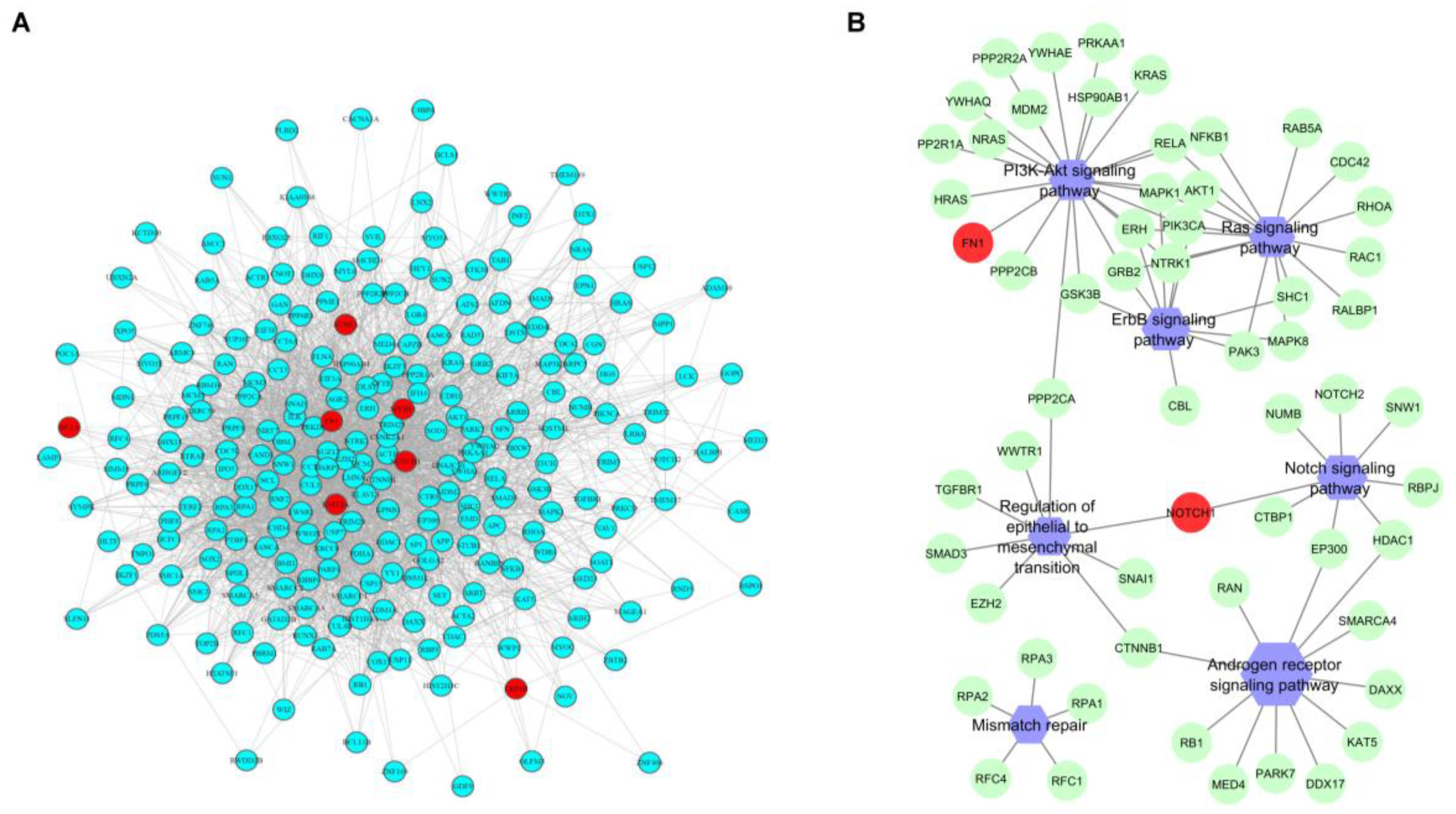
2.3. Genes and Pathways Associated with pCR
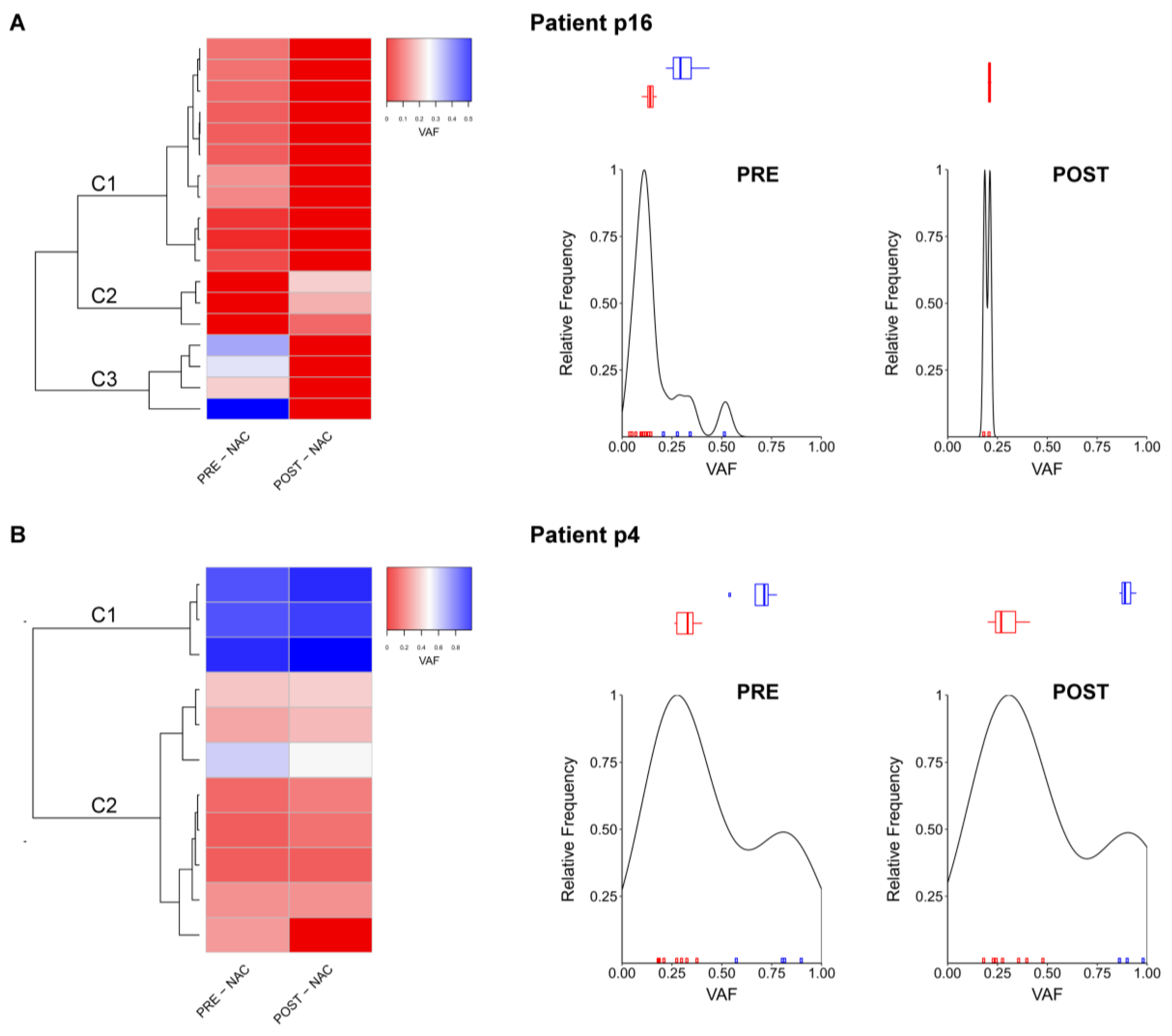
2.4. Comparison between Pre- and Post-NAC Samples
2.5. Tumor-Infiltrating Lymphocytes
3. Discussion
4. Materials and Methods
4.1. Patient Cohort
4.2. Targeted Next Generation Sequencing
4.3. Sequencing and Bio-Informatic Data Analysis
4.4. Statistical Analysis for Association of Genomic and Clinical Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16 (Suppl. 1), 1–11. [Google Scholar] [CrossRef]
- Mustacchi, G.; De Laurentiis, M. The role of taxanes in triple-negative breast cancer: Literature review. Drug Des. Dev. Ther. 2015, 9, 4303–4318. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, L.S.; Mayer, I.A. Platinum agents in the treatment of early-stage triple-negative breast cancer: Is it time to change practice? Clin. Adv. Hematol. Oncol. 2014, 12, 654–658. [Google Scholar] [PubMed]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [PubMed]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef]
- Masuda, N.; Lee, S.J.; Ohtani, S.; Im, Y.H.; Lee, E.S.; Yokota, I.; Kuroi, K.; Im, S.A.; Park, B.W.; Kim, S.B.; et al. Adjuvant Capecitabine for Breast Cancer after Preoperative Chemotherapy. N. Engl. J. Med. 2017, 376, 2147–2159. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef]
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2016, 13, 335–347. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlien, A.; Raine, K.; Fuligni, F.; Arnold, R.; Nik-Zainal, S.; Dronov, S.; Mamanova, L.; Rosic, A.; Ju, Y.S.; Cooke, S.L.; et al. Direct Transcriptional Consequences of Somatic Mutation in Breast Cancer. Cell Rep. 2016, 16, 2032–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertucci, F.; Chaffanet, M.; Birnbaum, D. An ICGC major achievement in breast cancer: A comprehensive catalog of mutations and mutational signatures. Chin. Clin. Oncol. 2017, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef]
- Lips, E.H.; Michaut, M.; Hoogstraat, M.; Mulder, L.; Besselink, N.J.; Koudijs, M.J.; Cuppen, E.; Voest, E.E.; Bernards, R.; Nederlof, P.M.; et al. Next generation sequencing of triple negative breast cancer to find predictors for chemotherapy response. Breast Cancer Res. 2015, 17, 134. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Shree, T.; Olson, O.C.; Elie, B.T.; Kester, J.C.; Garfall, A.L.; Simpson, K.; Bell-McGuinn, K.M.; Zabor, E.C.; Brogi, E.; Joyce, J.A. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011, 25, 2465–2479. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, B.L.; Anderson, R. Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy. Int. J. Mol. Sci. 2019, 20, 959. [Google Scholar] [CrossRef]
- Awasthee, N.; Rai, V.; Chava, S.; Nallasamy, P.; Kunnumakkara, A.B.; Bishayee, A.; Chauhan, S.C.; Challagundla, K.B.; Gupta, S.C. Targeting IκappaB kinases for cancer therapy. Semin Cancer Biol. 2019, 56, 12–24. [Google Scholar] [CrossRef]
- Banerjee, S.; Gusho, E.; Gaughan, C.; Dong, B.; Gu, X.; Holvey-Bates, E.; Talukdar, M.; Li, Y.; Weiss, S.R.; Sicheri, F.; et al. OAS-RNase L innate immune pathway mediates the cytotoxicity of a DNA-demethylating drug. Proc. Natl. Acad. Sci. USA 2019, 12, 5071–5076. [Google Scholar] [CrossRef] [PubMed]
- Martelotto, L.G.; Ng, C.K.Y.; Piscuoglio, S.; Weigelt, B.; Reis-Filho, J.S. Breast cancer intra-tumor heterogeneity. Breast Cancer Res. 2014, 16, 210. [Google Scholar] [CrossRef] [PubMed]
- Pribluda, A.; de la Cruz, C.C.; Jackson, E.L. Intratumoral Heterogeneity: From Diversity Comes Resistance. Clin. Cancer Res. 2015, 21, 2916–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieci, M.V.; Criscitiello, C.; Goubar, A.; Viale, G.; Conte, P.; Guarneri, V.; Ficarra, G.; Mathieu, M.C.; Delaloge, S.; Curigliano, G.; et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: A retrospective multicenter study. Ann. Oncol. 2014, 25, 611–618. [Google Scholar] [CrossRef]
- Luen, S.J.; Salagado, M.; Dieci, V.; Vingiani, A.; Curigliano, G.; Gould, R.E.; Castaneda, C.; D’Alfonso, T.; Sanchez, J.; Cheng, E.; et al. Prognostic implications of residual disease tumor-infiltrating lymphocytes and residual cancer burden in triple-negative breast cancer patients after neoadjuvant chemotherapy. Ann. Oncol. 2019, 30, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Baili, P.; Torresani, M.; Agresti, R.; Rosito, G.; Daidone, M.G.; Veneroni, S.; Cavallo, I.; Funaro, F.; Giunco, M.; Turco, A.; et al. A Breast Cancer Clinical Registry in An Italian Comprehensive Cancer Center: An Instrument for Descriptive, Clinical, and Experimental Research. Tumori. 2015, 101, 440–446. [Google Scholar] [CrossRef]
- Edge, S.B.; Compton, C.C. The American Joint Committee on Cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol. 2010, 17, 1471–1474. [Google Scholar] [CrossRef]
- Hammond, M.E.; Hayes, D.F.; Wolff, A.C.; Mangu, P.B.; Temin, S. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Oncol. Pract. 2010, 6, 195–197. [Google Scholar] [CrossRef]
- Wolff, A.C.; Hammond, M.E.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 2013, 25, 118–145. [Google Scholar] [CrossRef]
- Salgado, R.; Denkert, C.; Demaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Van den Eynden, G.; Baehner, F.L.; Penault-Llorca, F.; et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Feldman, L.D.; Hortobagyi, G.N.; Buzdar, A.U.; Ames, F.C.; Blumenschein, G.R. Pathological assessment of response to induction chemotherapy in breast cancer. Cancer Res. 1986, 46, 2578–2581. [Google Scholar] [PubMed]
- Ramos, A.H.; Lichtenstein, L.; Gupta, M.; Lawrence, M.S.; Pugh, T.J.; Saksena, G.; Meyerson, M.; Getz, G. Oncotator: Cancer variant annotation tool. Hum. Mutat. 2015, 36, E2423–E2429. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Diez, D.; Hutchins, A.P.; Miranda-Saavedra, D. Systematic identification of transcriptional regulatory modules from protein-protein interaction networks. Nucleic Acids Res. 2014, 42, e6. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | N (%) |
|---|---|
| Patient age | |
| <50 years | 12 (63) |
| ≥50 years | 7 (37) |
| Clinical T size | |
| 2–5 cm | 13 (68) |
| >5 cm | 6 (32) |
| Clinical nodal status | |
| N0 | 8 (42) |
| N1-3 | 11 (58) |
| Clinical stage | |
| IIA | 5 (26) |
| IIB | 10 (53) |
| IIIA | 2 (11) |
| IIIB | 1 (5) |
| IIIC * | 1 (5) |
| Tumor grade | |
| G2 | 1 (5) |
| G3 | 18 (95) |
| Ki67 | |
| <30% | 3 (16) |
| ≥30% <60% | 3 (16) |
| ≥60% | 12 (63) |
| Missing | 1 (5) |
| Type of NAC | |
| – Doxorubicin/Paclitaxel every 3 weeks followed by CMF1–8 every 4 weeks ^ | 11 (58) |
| – Doxorubicin/Paclitaxel every 3 weeks followed by Eribulin 1–8 every 3 weeks ^^ | 5 (26) |
| – Other | 3 (16) |
| Path Findings | |
| pCR | |
| ypT0N0 | 4 (21) |
| Residual Disease | |
| ypT1N0 | 6 (32) |
| ypT2-3N0 | 7 (37) |
| ypT2N1 | 1 (5) |
| ypT4N3 | 1 (5) |
| Evaluable Cases | |
| Pre-NAC | 16 (84) |
| Post-NAC | 15 (79) |
| Paired pre- and post-NAC | 12 (63) |
| Events | |
| Distant metastases | 7 (37) |
| Patient | Cluster | No. of Genes in Cluster | No. of Genes in Cluster Mutated in POST-NAC Tumor | Enriched Pathways Including Genes Mutated in POST-NAC Tumor |
|---|---|---|---|---|
| p5 | C1 | 42 | 1 | No pathways found |
| C2 | 10 | 1 | No pathways found | |
| C3 | 15 | 0 | NA | |
| p10 | C1 | 3 | 3 | EGFR tyrosine kinase inhibitor resistance (KEGG - hsa01521); Ras signaling pathway (KEGG - hsa04014) |
| C2 | 3 | 0 | NA | |
| p11 | C1 | 3 | 2 | Negative regulation of cell cycle process (GO:0010948); Androgen receptor signaling pathway (GO:0030521) |
| C2 | 5 | 2 | Ras signaling pathway (hsa04014); TOR signaling (GO:0031929); EGFR tyrosine kinase inhibitor resistance (KEGG - hsa01521) | |
| p12 | C1 | 3 | 3 | mTOR signaling pathway (KEGG - hsa04150); PI3K-Akt signaling pathway (KEGG - hsa04151); |
| C2 | 2 | 0 | NA | |
| p13 | C1 | 1 | 1 | PI3K-Akt signaling pathway (KEGG - hsa04151); Negative regulation of cell cycle process (GO:0010948) |
| C2 | 4 | 2 | TORC1 signaling (GO:0038202) | |
| p16 | C1 | 11 | 0 | NA |
| C2 | 3 | 3 | Hippo signaling pathway (KEGG - hsa04390) | |
| C3 | 4 | 0 | NA | |
| p18 | C1 | 2 | 2 | Regulation of cell cycle arrest (GO:0071156); PI3K-Akt signaling pathway (KEGG - hsa04151) |
| C2 | 2 | 2 | Mismatch repair (KEGG - hsa03430); Platinum drug resistance (KEGG - hsa01524) | |
| C3 | 2 | 0 | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Cosimo, S.; Appierto, V.; Silvestri, M.; Pruneri, G.; Vingiani, A.; Perrone, F.; Busico, A.; Folli, S.; Scaperrotta, G.; de Braud, F.G.; et al. Targeted-Gene Sequencing to Catch Triple Negative Breast Cancer Heterogeneity before and after Neoadjuvant Chemotherapy. Cancers 2019, 11, 1753. https://doi.org/10.3390/cancers11111753
Di Cosimo S, Appierto V, Silvestri M, Pruneri G, Vingiani A, Perrone F, Busico A, Folli S, Scaperrotta G, de Braud FG, et al. Targeted-Gene Sequencing to Catch Triple Negative Breast Cancer Heterogeneity before and after Neoadjuvant Chemotherapy. Cancers. 2019; 11(11):1753. https://doi.org/10.3390/cancers11111753
Chicago/Turabian StyleDi Cosimo, Serena, Valentina Appierto, Marco Silvestri, Giancarlo Pruneri, Andrea Vingiani, Federica Perrone, Adele Busico, Secondo Folli, Gianfranco Scaperrotta, Filippo Guglielmo de Braud, and et al. 2019. "Targeted-Gene Sequencing to Catch Triple Negative Breast Cancer Heterogeneity before and after Neoadjuvant Chemotherapy" Cancers 11, no. 11: 1753. https://doi.org/10.3390/cancers11111753