Coupled Genome-Wide DNA Methylation and Transcription Analysis Identified Rich Biomarkers and Drug Targets in Triple-Negative Breast Cancer

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Datasets Used in the Study
2.2. Analysis of DNA Methylation Data
2.3. Analysis of Gene Expression Data
2.4. Analysis of DMGs and DEGs in Different Regions
2.5. Functional Enrichment Analysis
2.6. Evaluation of Methylation and Expression Biomarkers
2.7. Identification of Potential Drug Targets
3. Results
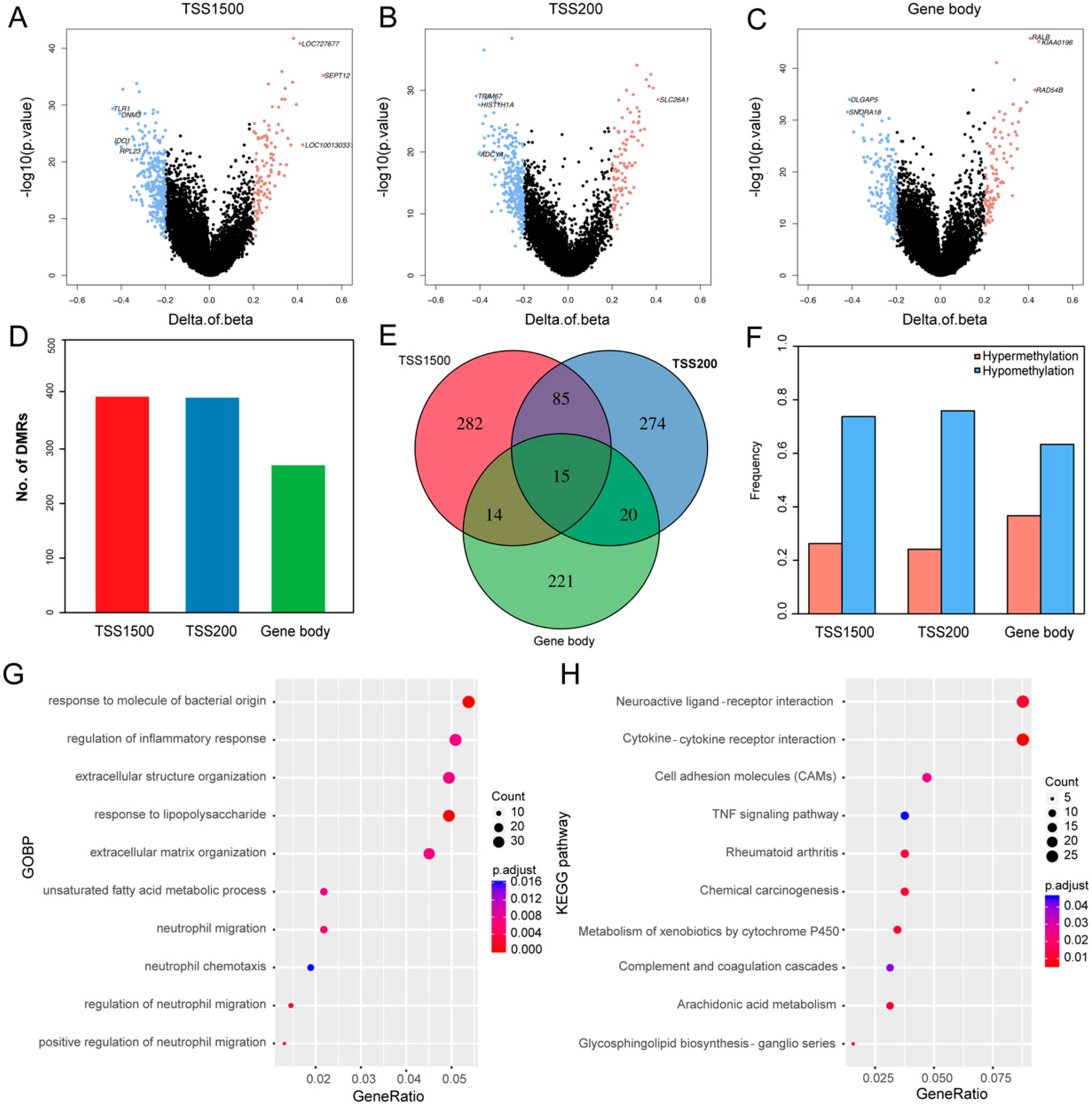
3.1. Differentially Methylated Genes (DMGs) in TNBC
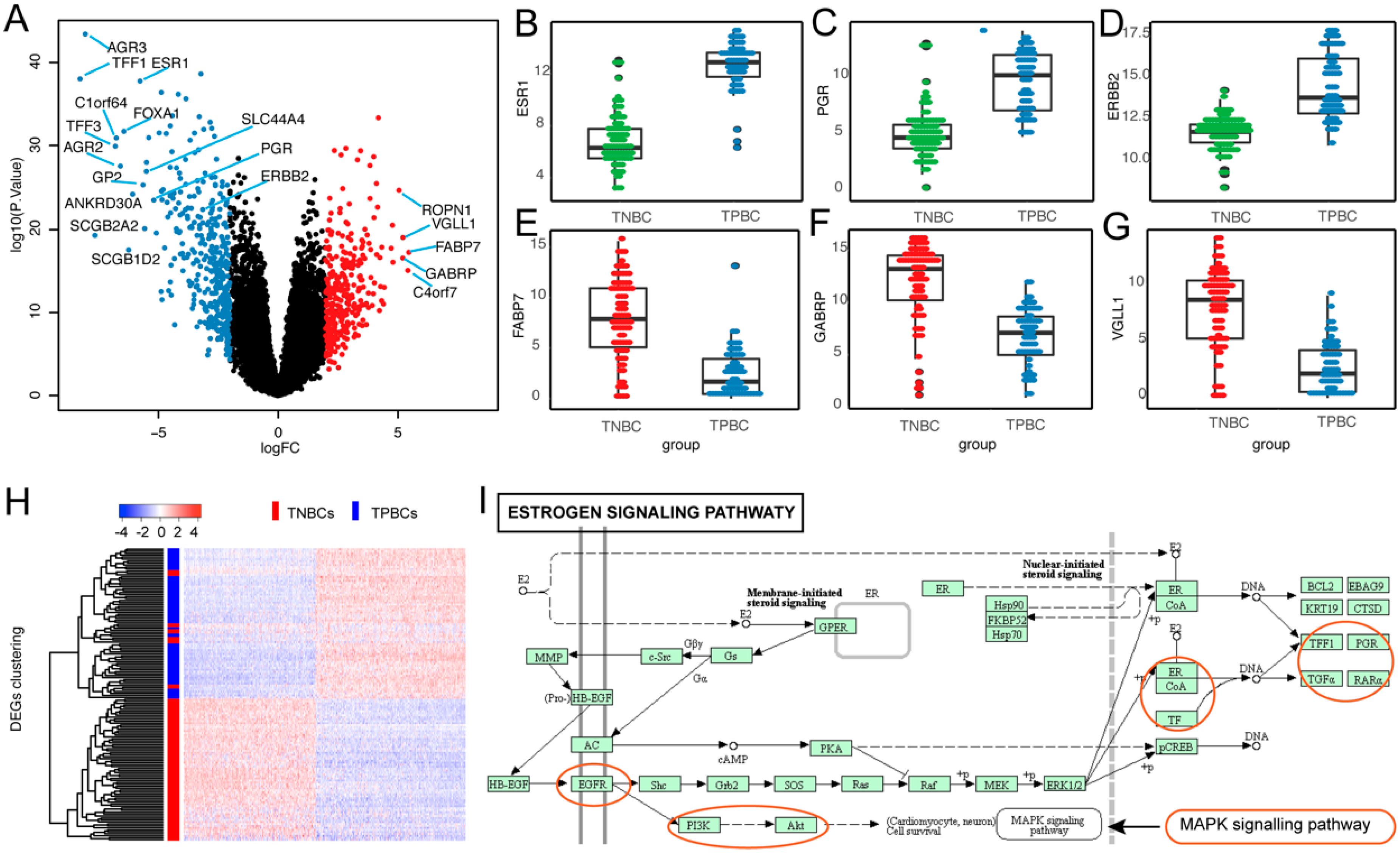
3.2. Differentially Expressed Genes (DEGs) in TNBC
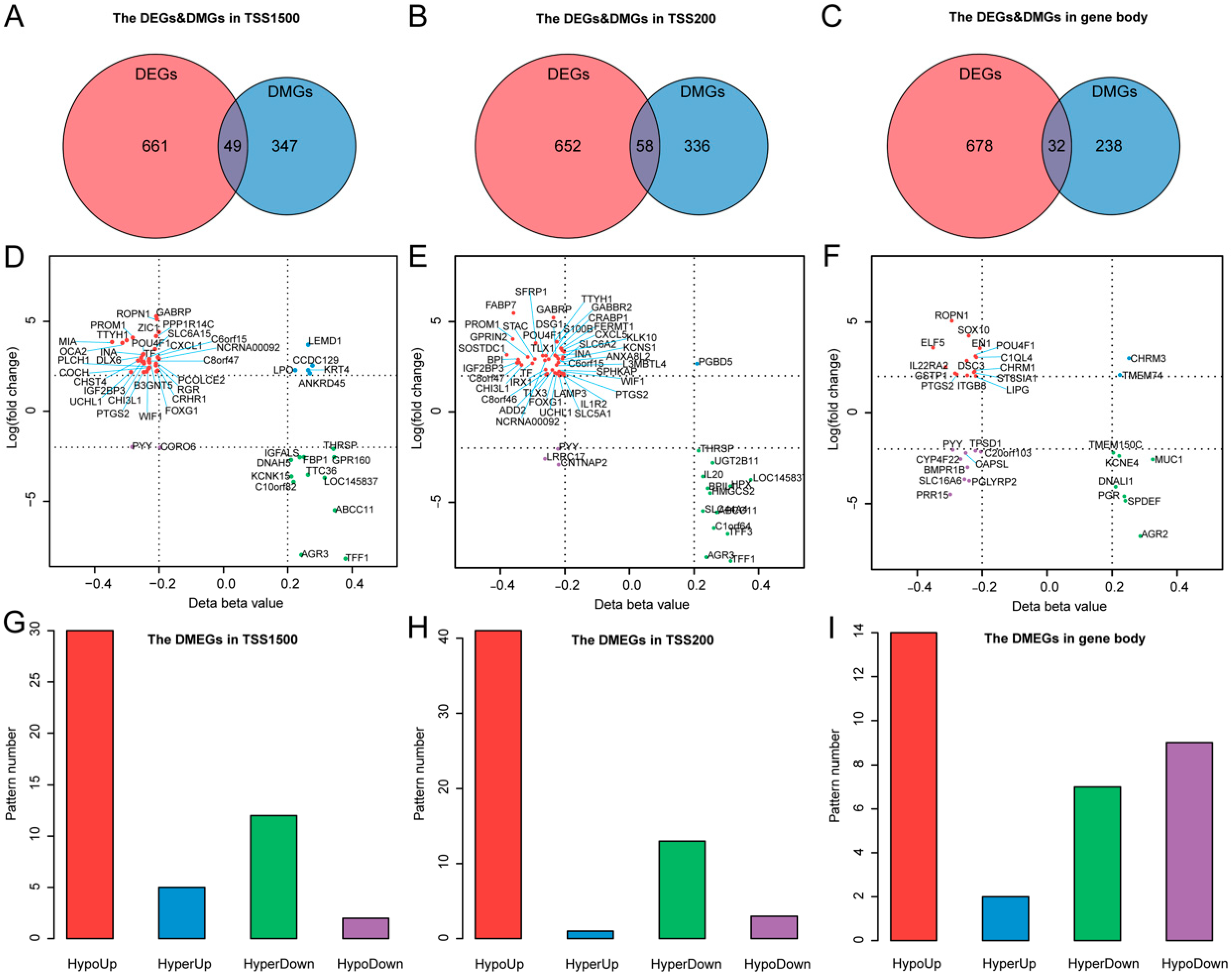
3.3. Differentially Methylated and Expressed Genes (DMEGs) in TNBC
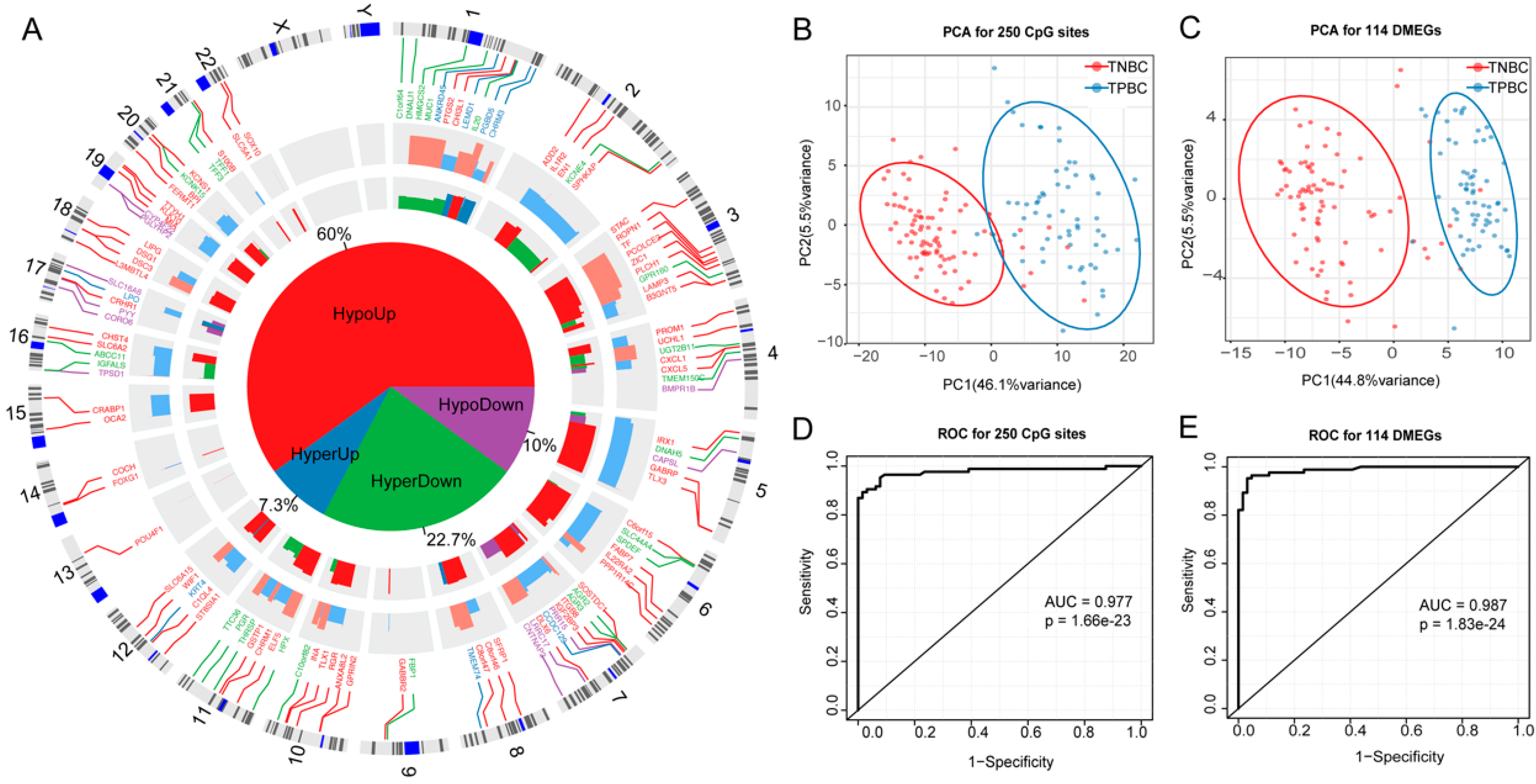
3.4. DMEGs Predicting TNBCs
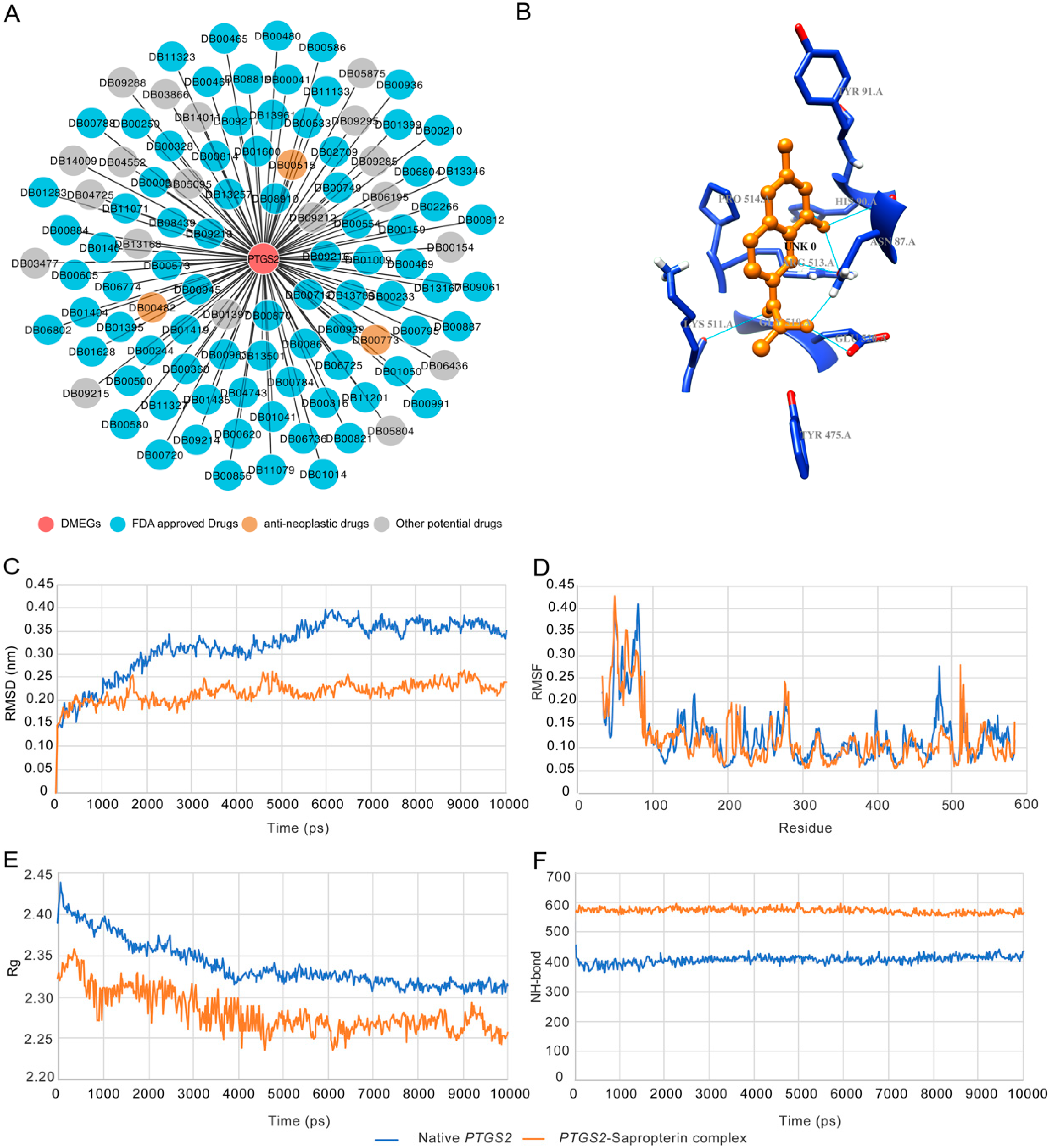
3.5. Multiple DMEGs are Potential Druggable Targets
3.6. Structural Basis of Sapropterin Bound to PTGS2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2011, 16 (Suppl. 1), 61–70. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Shimelis, H.; LaDuca, H.; Hu, C.; Hart, S.N.; Na, J.; Thomas, A.; Akinhanmi, M.; Moore, R.M.; Brauch, H.; Cox, A.; et al. Triple-negative breast cancer risk genes identified by multigene hereditary cancer panel testing. J. Natl. Cancer Inst. 2018, 110, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, H.G.; Malmgren, J.A.; Atwood, M. T1N0 triple negative breast cancer: Risk of recurrence and adjuvant chemotherapy. Breast J. 2009, 15, 454–460. [Google Scholar] [CrossRef]
- Tan, A.R.; Swain, S.M. Therapeutic strategies for triple-negative breast cancer. Cancer J. 2008, 14, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Mersin, H.; Yildirim, E.; Berberoglu, U.; Gulben, K. The prognostic importance of triple negative breast carcinoma. Breast 2008, 17, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Good, C.R.; Panjarian, S.; Kelly, A.D.; Madzo, J.; Patel, B.; Jelinek, J.; Issa, J.J. TET1-mediated hypomethylation activates oncogenic signaling in triple-negative breast cancer. Cancer Res. 2018, 78, 4126–4137. [Google Scholar] [CrossRef]
- Shin, E.; Lee, Y.; Koo, J.S. Differential expression of the epigenetic methylation-related protein DNMT1 by breast cancer molecular subtype and stromal histology. J. Transl. Med. 2016, 14, 87. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.; Yu, H.; Tian, J.; Yuan, F.; Fan, J.; Liu, Y.; Zhu, L.; Wang, F.; Zhao, Y.; et al. DUSP1 promoter methylation in peripheral blood leukocyte is associated with triple-negative breast cancer risk. Sci. Rep. 2017, 7, 43011. [Google Scholar] [CrossRef]
- Yamashita, N.; Tokunaga, E.; Kitao, H.; Hitchins, M.; Inoue, Y.; Tanaka, K.; Hisamatsu, Y.; Taketani, K.; Akiyoshi, S.; Okada, S.; et al. Epigenetic inactivation of BRCA1 through promoter hypermethylation and its clinical importance in triple-negative breast cancer. Clin. Breast Cancer 2015, 15, 498–504. [Google Scholar] [CrossRef]
- Mathe, A.; Wong-Brown, M.; Locke, W.J.; Stirzaker, C.; Braye, S.G.; Forbes, J.F.; Clark, S.J.; Avery-Kiejda, K.A.; Scott, R.J. DNA methylation profile of triple negative breast cancer-specific genes comparing lymph node positive patients to lymph node negative patients. Sci. Rep. 2016, 6, 33435. [Google Scholar] [CrossRef] [PubMed]
- Stirzaker, C.; Zotenko, E.; Song, J.Z.; Qu, W.; Nair, S.S.; Locke, W.J.; Stone, A.; Armstong, N.J.; Robinson, M.D.; Dobrovic, A.; et al. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat. Commun. 2015, 6, 5899. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Yao, J.; Li, P.; Zhang, C.; Lowery, F.J.; Zhang, Q.; Guo, H.; Qu, J.; Yang, F.; Wistuba, I.I.; et al. Sphingosine-kinase-1 signaling promotes metastasis of triple-negative breast cancer. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Peluffo, G.; Subedee, A.; Harper, N.W.; Kingston, N.; Jovanovic, B.; Flores, F.; Stevens, L.E.; Beca, F.; Trinh, A.; Chilamakuri, C.S.R.; et al. EN1 is a transcriptional dependency in triple-negative breast cancer associated with brain metastasis. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Song, L.; Xu, Y.; Wu, Y.; Dai, C.; Wang, X.; Sun, X.; Hou, Y.; Li, W.; Zhan, X.; et al. Loss of Wwox drives metastasis in triple-negative breast cancer by JAK2/STAT3 axis. Nat. Commun. 2018, 9, 3486. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; Kamath, A.; McDade, F.; Rogers, D.; Brooks, A.N.; Zhu, J.; Haussler, D. The UCSC Xena Platform for cancer genomics data visualization and interpretation. BioRxiv 2019. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- Du, P.; Kibbe, W.A.; Lin, S.M. lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Quintero, M.; Adamoski, D.; Reis, L.M.D.; Ascencao, C.F.R.; Oliveira, K.R.S.; Goncalves, K.A.; Dias, M.M.; Carazzolle, M.F.; Dias, S.M.G. Guanylate-binding protein-1 is a potential new therapeutic target for triple-negative breast cancer. BMC Cancer 2017, 17, 727. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, G.M.; Sizemore, S.T.; Seachrist, D.D.; Keri, R.A. GABA(A) receptor pi (GABRP) stimulates basal-like breast cancer cell migration through activation of extracellular-regulated kinase 1/2 (ERK1/2). J. Biol. Chem. 2014, 289, 24102–24113. [Google Scholar] [CrossRef] [PubMed]
- Castilla, M.A.; Lopez-Garcia, M.A.; Atienza, M.R.; Rosa-Rosa, J.M.; Diaz-Martin, J.; Pecero, M.L.; Vieites, B.; Romero-Perez, L.; Benitez, J.; Calcabrini, A.; et al. VGLL1 expression is associated with a triple-negative basal-like phenotype in breast cancer. Endocr. Relat. Cancer 2014, 21, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.Z.; Graham, K.; Glubrecht, D.D.; Lai, R.; Mackey, J.R.; Godbout, R. A fatty acid-binding protein 7/RXRβ pathway enhances survival and proliferation in triple-negative breast cancer. J. Pathol. 2012, 228, 310–321. [Google Scholar] [CrossRef]
- Ignacio, R.M.C.; Gibbs, C.R.; Lee, E.S.; Son, D.S. The TGFα-EGFR-Akt signaling axis plays a role in enhancing proinflammatory chemokines in triple-negative breast cancer cells. Oncotarget 2018, 9, 29286–29303. [Google Scholar] [CrossRef]
- Omata, F.; McNamara, K.M.; Suzuki, K.; Abe, E.; Hirakawa, H.; Ishida, T.; Ohuchi, N.; Sasano, H. Effect of the normal mammary differentiation regulator ELF5 upon clinical outcomes of triple negative breast cancers patients. Breast Cancer 2018, 25, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.K.; Yao, Y.; Lee, J.S.; Zhang, Y.; Huang, W.; Kane, M.A.; Zhou, Q. LIPG signaling promotes tumor initiation and metastasis of human basal-like triple-negative breast cancer. Elife 2018, 7, e31334. [Google Scholar] [CrossRef]
- Lien, H.C.; Wang, C.C.; Lin, C.H.; Lu, Y.S.; Huang, C.S.; Hsiao, L.P.; Yao, Y.T. Differential expression of ubiquitin carboxy-terminal hydrolase L1 in breast carcinoma and its biological significance. Hum. Pathol. 2013, 44, 1838–1848. [Google Scholar] [CrossRef]
- Jezequel, P.; Loussouarn, D.; Guerin-Charbonnel, C.; Campion, L.; Vanier, A.; Gouraud, W.; Lasla, H.; Guette, C.; Valo, I.; Verriele, V.; et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: Importance of immune response. Breast Cancer Res. 2015, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Hachim, M.Y.; Hachim, I.Y.; Dai, M.; Lo, C.; Raffa, F.A.; Ali, S.; Lebrun, J.J. Cyclooxygenase-2 regulates TGFβ-induced cancer stemness in triple-negative breast cancer. Sci. Rep. 2017, 7, 40258. [Google Scholar] [CrossRef]
- Kochel, T.J.; Goloubeva, O.G.; Fulton, A.M. Upregulation of cyclooxygenase-2/prostaglandin E2 (COX-2/PGE2) pathway member multiple drug resistance-associated protein 4 (MRP4) and downregulation of prostaglandin transporter (PGT) and 15-prostaglandin dehydrogenase (15-PGDH) in triple-negative breast cancer. Breast Cancer 2016, 10, 61–70. [Google Scholar] [CrossRef]
- Alikanoglu, A.S.; Yildirim, M.; Suren, D.; Yildiz, M.; Kaya, V.; Dilli, U.D.; Sezer, C. Expression of cyclooxygenase-2 and Bcl-2 in breast cancer and their relationship with triple-negative disease. J. BUON 2014, 19, 430–434. [Google Scholar]
- Chen, G.; Zhang, H.; Sun, L.; Jiang, Y.; Xu, Z.; Gu, H.; Xu, H.; Yang, J.; Wang, Y.; Xu, T.; et al. Prognostic significance of GSTP1 in patients with triple negative breast cancer. Oncotarget 2017, 8, 68675–68680. [Google Scholar] [CrossRef]
- Louie, S.M.; Grossman, E.A.; Crawford, L.A.; Ding, L.; Camarda, R.; Huffman, T.R.; Miyamoto, D.K.; Goga, A.; Weerapana, E.; Nomura, D.K. GSTP1 is a driver of triple-negative breast cancer cell metabolism and pathogenicity. Cell Chem. Biol. 2016, 23, 567–578. [Google Scholar] [CrossRef]
- Zafrakas, M.; Chorovicer, M.; Klaman, I.; Kristiansen, G.; Wild, P.J.; Heindrichs, U.; Knuchel, R.; Dahl, E. Systematic characterisation of GABRP expression in sporadic breast cancer and normal breast tissue. Int. J. Cancer 2006, 118, 1453–1459. [Google Scholar] [CrossRef]
- Naves, M.A.; Graminha, A.E.; Vegas, L.C.; Luna-Dulcey, L.; Honorato, J.; Menezes, A.C.S.; Batista, A.A.; Cominetti, M.R. Transport of the ruthenium complex [Ru(GA)(dppe)2]PF6 into triple-negative breast cancer cells is facilitated by transferrin receptors. Mol. Pharm. 2019, 16, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Henry, K.E.; Dilling, T.R.; Abdel-Atti, D.; Edwards, K.J.; Evans, M.J.; Lewis, J.S. Noninvasive (89)Zr-transferrin PET shows improved tumor targeting compared with (18)F-FDG PET in MYC-overexpressing human triple-negative breast cancer. J. Nucl. Med. 2018, 59, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.C.; Huang, Y.C.; Kan, J.Y.; Kuo, P.L.; Hou, M.F.; Hsu, Y.L. S100B expression in breast cancer as a predictive marker for cancer metastasis. Int. J. Oncol. 2018, 52, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, T.; Madsen, J.S.; Brandslund, I.; Lund, E.D.; Ormstrup, T.; Jakobsen, E.H.; Jylling, A.M.; Steffensen, K.D.; Jakobsen, A. Predicting brain metastases of breast cancer based on serum S100B and serum HER2. Oncol. Lett. 2013, 6, 1265–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.T.; Chen, T.M.; Mei, C.T.; Chang, C.F.; Liu, L.L.; Chiu, K.H.; Wu, T.M.; Lan, Y.C.; Liu, W.S.; Chen, Y.H.; et al. The functional haplotypes of CHRM3 modulate mRNA expression and associate with bladder cancer among a Chinese Han population in Kaohsiung City. Biomed. Res. Int. 2016, 2016, 4052846. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Wen, S.; Yang, X.; Zhang, Y.; Wang, Z.; Zhang, Z. CHRM3 is a novel prognostic factor of poor prognosis in patients with endometrial carcinoma. Am. J. Transl. Res. 2015, 7, 902–911. [Google Scholar]
- Wang, N.; Yao, M.; Xu, J.; Quan, Y.; Zhang, K.; Yang, R.; Gao, W.Q. Autocrine activation of CHRM3 promotes prostate cancer growth and castration resistance via CaM/CaMKK-mediated phosphorylation of Akt. Clin. Cancer Res. 2015, 21, 4676–4685. [Google Scholar] [CrossRef]
- Padhye, S.; Ahmad, A.; Oswal, N.; Dandawate, P.; Rub, R.A.; Deshpande, J.; Swamy, K.V.; Sarkar, F.H. Fluorinated 2′-hydroxychalcones as garcinol analogs with enhanced antioxidant and anticancer activities. Bioorganic Med. Chem. Lett. 2010, 20, 5818–5821. [Google Scholar] [CrossRef]
- Youn, A.; Kim, K.I.; Rabadan, R.; Tycko, B.; Shen, Y.; Wang, S. A pan-cancer analysis of driver gene mutations, DNA methylation and gene expressions reveals that chromatin remodeling is a major mechanism inducing global changes in cancer epigenomes. BMC Med. Genom. 2018, 11, 98. [Google Scholar] [CrossRef]
- Gao, Y.; Teschendorff, A.E. Epigenetic and genetic deregulation in cancer target distinct signaling pathway domains. Nucleic Acids Res. 2017, 45, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Jones, A.; Widschwendter, M.; Teschendorff, A.E. An integrative pan-cancer-wide analysis of epigenetic enzymes reveals universal patterns of epigenomic deregulation in cancer. Genome Biol. 2015, 16, 140. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, L.; Shu, X.O.; Cai, Q.; Shu, X.; Li, B.; Guo, X.; Ye, F.; Michailidou, K.; Bolla, M.K.; et al. Genetically predicted levels of DNA methylation biomarkers and breast cancer risk: Data from 228,951 women of European descent. J. Natl. Cancer Inst. 2019. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gonzalez, A.; Munoz-Muela, E.; Marchal, J.A.; Cara, F.E.; Molina, M.P.; Cruz-Lozano, M.; Jimenez, G.; Verma, A.; Ramirez, A.; Qian, W.; et al. Activating transcription factor 4 modulates TGFβ-induced aggressiveness in triple-negative breast cancer via SMAD2/3/4 and mTORC2 signaling. Clin. Cancer Res. 2018, 24, 5697–5709. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, S.; Fukushima, W.; Asano, Y.; Goto, W.; Takada, K.; Noda, S.; Takashima, T.; Onoda, N.; Ohsawa, M.; Hirakawa, K.; et al. Identification of predictive markers of the therapeutic effect of eribulin chemotherapy for locally advanced or metastatic breast cancer. BMC Cancer 2017, 17, 604. [Google Scholar] [CrossRef]
- Hsia, T.C.; Tu, C.Y.; Chen, Y.J.; Wei, Y.L.; Yu, M.C.; Hsu, S.C.; Tsai, S.L.; Chen, W.S.; Yeh, M.H.; Yen, C.J.; et al. Lapatinib-mediated cyclooxygenase-2 expression via epidermal growth factor receptor/HuR interaction enhances the aggressiveness of triple-negative breast cancer cells. Mol. Pharmacol. 2013, 83, 857–869. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Methylation Cut-Off | Expression Cut-Off |
|---|---|---|
| HypoUp | adjusted p-value < 0.05 and delta β-value < −0.2 | adjusted p value <0.05 and logFC > 2 |
| HypoDown | adjusted p-value < 0.05 and delta β-value < −0.2 | adjusted p value <0.05 and logFC < −2 |
| HyperUp | adjusted p-value < 0.05 and delta β-value > 0.2 | adjusted p value <0.05 and logFC > 2 |
| HyperDown | adjusted p-value < 0.05 and delta β-value > 0.2 | adjusted p value <0.05 and logFC < −2 |
| Type | RefGene |
|---|---|
| Receptors | GPR160, GABRP, RGR, CRHR1, GABBR2, IL1R2, PGR, CHRM3, IL22RA2, BMPR1B, CHRM1 |
| Functional Proteins | AGR3, PPP1R14C, ROPN1, OCA2, IGFALS, IGF2BP3, INA, L3MBTL4, SFRP1, FABP7, GPRIN2, S100B, LAMP3, BPI, CNTNAP2, CRABP1, AGR2, PGLYRP2, TMEM74, TMEM150C |
| Structural Proteins | TFF1, B3GNT5, KCNK15, LOC145837, FBP1, NCRNA00092, TTC36, ABCC11, LEMD1, C10orf82, ZIC1, C8orf47, POU4F1, DNAH5, CHST4, TTYH1, CXCL1, PROM1, DLX6, ANKRD45, MIA, PYY, LPO, PCOLCE2, CHI3L1, C6orf15, UCHL1, CCDC129, PLCH1, TF, SLC6A15, CORO6, FOXG1, COCH, PTGS2, KRT4, THRSP, WIF1, C1orf64, TFF3, SLC44A4, HPX, STAC, BPIL1, FERMT1, PGBD5, IL20, LRRC17, SLC6A2, KCNS1, CXCL5, HMGCS2, ADD2, SOSTDC1, TLX1, TLX3, KLK10, SPHKAP, DSG1, ANXA8L2, IRX1, UGT2B11, C8orf46, SLC5A1, PRR15, SLC16A6, DNALI1, GSTP1, SPDEF, ST8SIA1, C1QL4, EN1, KCNE4, MUC1, C20orf103, ITGB8, CAPSL, ELF5, LIPG, SOX10, DSC3, CYP4F22, TPSD1 |
| RefGene | Region | CpG Sites | DMS | Pattern | Drugs | Drug Examples |
|---|---|---|---|---|---|---|
| BPI | TSS200 | 3 | 1 | HypoUp | 1 | Fostamatinib |
| CHRM1 | Body | 3 | 2 | HypoUp | 95 | Cevimeline, tramadol, succinylcholine |
| CHRM3 | Body | 8 | 6 | HyperUp | 75 | Ziprasidone, disopyramide, ipratropium |
| GABRP | TSS200 | 3 | 2 | HypoUp | 16 | Prazepam, quazepam, nitrazepam |
| TSS1500 | 3 | 1 | HypoUp | 16 | Prazepam, quazepam, nitrazepam | |
| GSTP1 | Body | 6 | 4 | HypoUp | 37 | Chlorambucil, cisplatin, busulfan |
| PTGS2 | TSS200 | 3 | 3 | HypoUp | 111 | Bufexamac, bendazac, acemetacin |
| TSS1500 | 6 | 4 | HypoUp | 111 | Bufexamac, bendazac, acemetacin | |
| Body | 5 | 2 | HypoUp | 111 | Bufexamac, bendazac, acemetacin | |
| S100B | TSS200 | 2 | 1 | HypoUp | 9 | Olopatadine, calcium, calcium citrate |
| SLC6A2 | TSS200 | 3 | 3 | HypoUp | 76 | Amphetamine, phentermine, tramadol |
| TF | TSS200 | 3 | 1 | HypoUp | 32 | Cisplatin, isoflurophate, iron dextran |
| TSS1500 | 4 | 2 | HypoUp | 32 | Cisplatin, isoflurophate, iron dextran | |
| UCHL1 | TSS200 | 2 | 1 | HypoUp | 1 | Phenethyl isothiocyanate |
| No. | FDA Drug | Binding Energy (kcal/mol) | No. of Hydrogen Bonds | Residues |
|---|---|---|---|---|
| 1 | Icosapent | −6.63 | 0 | - |
| 2 | Adapalene | −6.99 | 1 | Gln-192 |
| 3 | Mesalazine | −6.84 | 1 | Val-523 |
| 4 | Dapsone | −6.48 | 2 | Gln-192, Ser-530 |
| 5 | Sapropterin | −6.15 | 6 | Asn-87 (3), His-90, Lys-511, Glu-520 |
| 6 | Flurbiprofen | −5.88 | 1 | Gln-192 |
| 7 | Ketorolac | −5.12 | 2 | Arg-513, Val-523 |
| 8 | Piroxicam | −4.71 | 2 | Ile-517 (2) |
| 9 | Phenylbutazone | −4.28 | 1 | Arg-513 |
| 10 | Mefenamic acid | −3.55 | 2 | Met-522, Gln-192 |
| 11 | Carprofen | −3.19 | 1 | Val-523 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, M.; Sinha, S.; Wang, S.M. Coupled Genome-Wide DNA Methylation and Transcription Analysis Identified Rich Biomarkers and Drug Targets in Triple-Negative Breast Cancer. Cancers 2019, 11, 1724. https://doi.org/10.3390/cancers11111724
Guo M, Sinha S, Wang SM. Coupled Genome-Wide DNA Methylation and Transcription Analysis Identified Rich Biomarkers and Drug Targets in Triple-Negative Breast Cancer. Cancers. 2019; 11(11):1724. https://doi.org/10.3390/cancers11111724
Chicago/Turabian StyleGuo, Maoni, Siddharth Sinha, and San Ming Wang. 2019. "Coupled Genome-Wide DNA Methylation and Transcription Analysis Identified Rich Biomarkers and Drug Targets in Triple-Negative Breast Cancer" Cancers 11, no. 11: 1724. https://doi.org/10.3390/cancers11111724