NF-κB Signaling in Targeting Tumor Cells by Oncolytic Viruses—Therapeutic Perspectives


Abstract
:1. Introduction
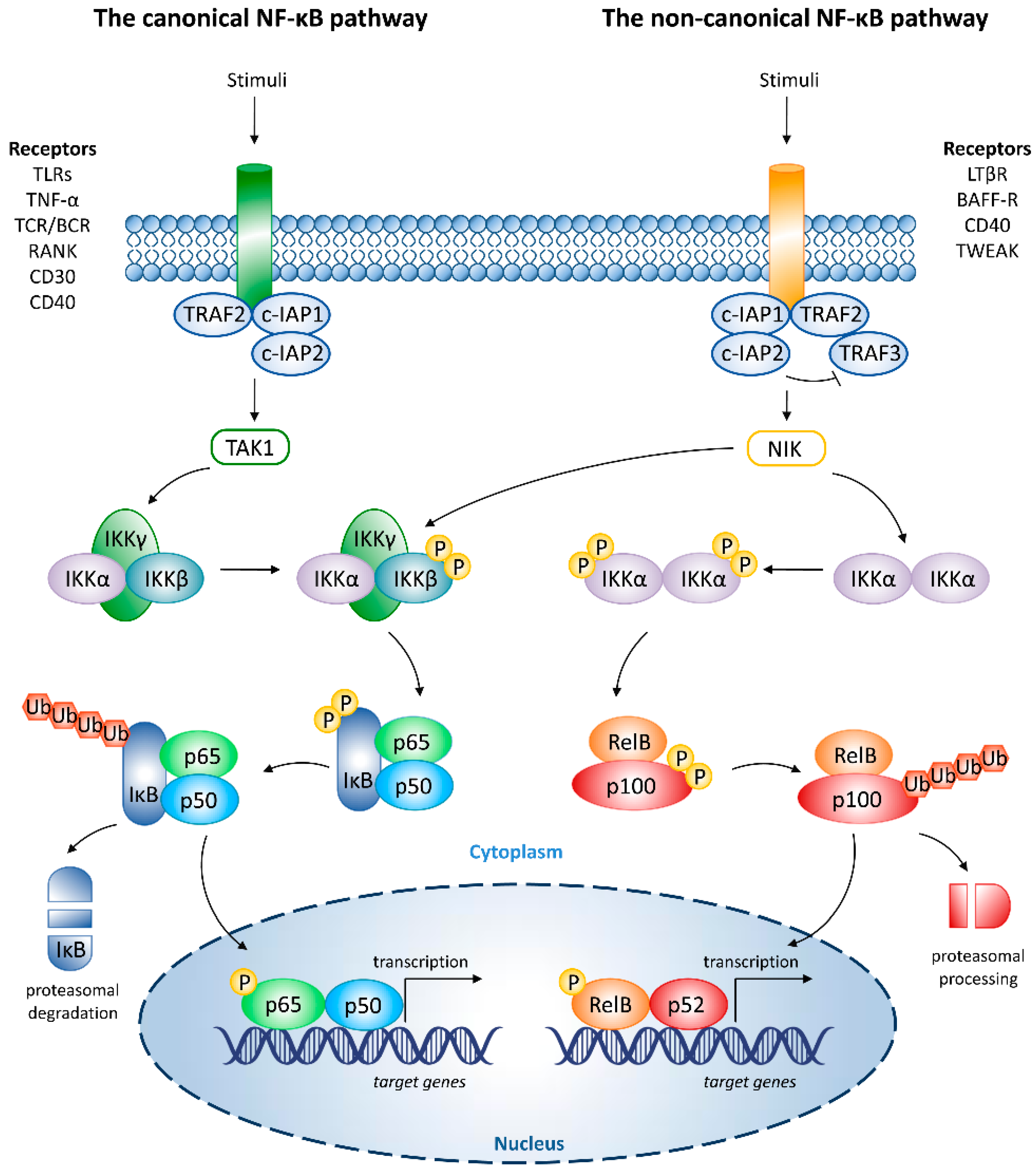
2. Overview of NF-κB Signaling
3. NF-κB in Oncogenesis
4. OVs
5. OVs and NF-κB
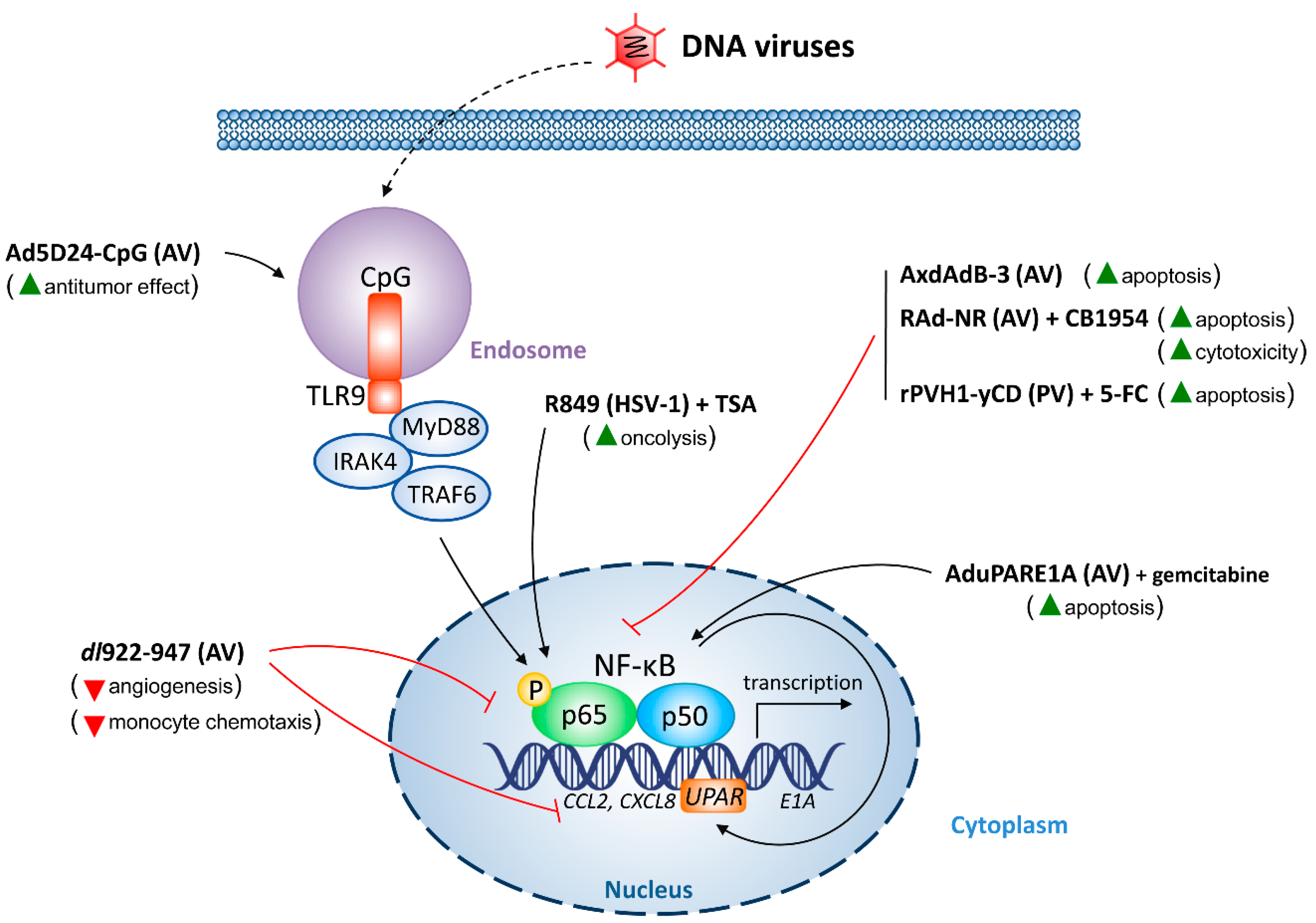
5.1. DNA Viruses
5.1.1. AV
5.1.2. HSV
5.1.3. PV
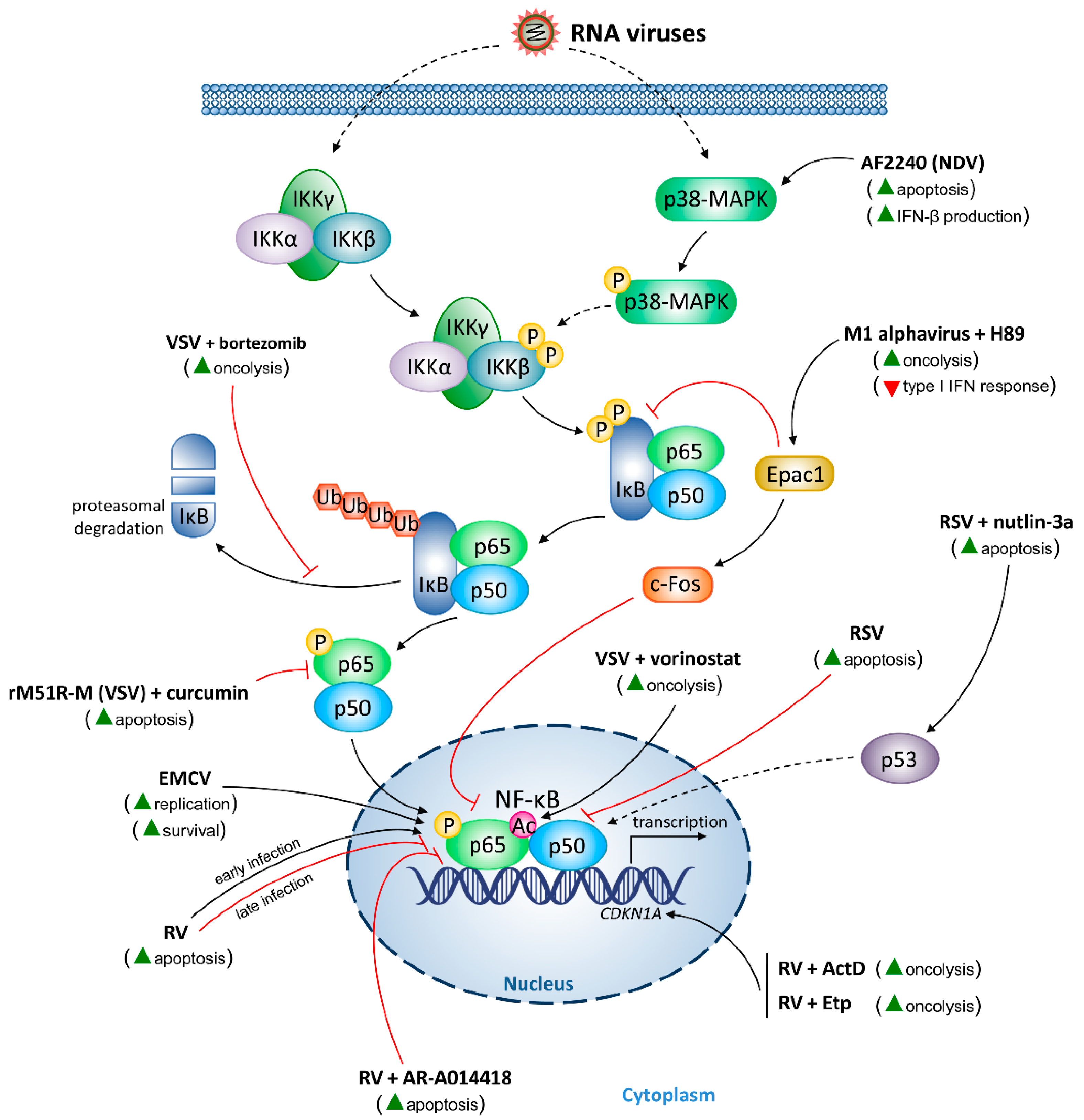
5.2. RNA Viruses
5.2.1. Encephalomyocarditis Virus (EMCV)
5.2.2. M1 Alphavirus
5.2.3. Newcastle Disease Virus (NDV)
5.2.4. RV
5.2.5. RSV
5.2.6. VSV
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jing, H.; Lee, S. NF-κB in cellular senescence and cancer treatment. Mol. Cells 2014, 37, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. NF-κB in cancer therapy. Arch. Toxicol. 2015, 89, 711–731. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.B.; Silva, R.C.M.C.; Ferreira, G.M.; Abdelhay, E. NF-κB: Two sides of the same coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, L.; Bigas, A.; Mulero, M.C. Alternative nuclear functions for NF-κB family members. Am. J. Cancer Res. 2011, 1, 446–459. [Google Scholar] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.-S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.; Yamazaki, S.; He, J.Q.; Cheng, G. Control of canonical NF-κB activation through the NIK-IKK complex pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 3503–3508. [Google Scholar] [CrossRef] [PubMed]
- Zeligs, K.P.; Neuman, M.K.; Annunziata, C.M. Molecular pathways: The balance between cancer and the immune system challenges the therapeutic specificity of targeting nuclear factor-κB signaling for cancer treatment. Clin. Cancer Res. 2016, 22, 4302–4308. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Geismann, C.; Erhart, W.; Grohmann, F.; Schreiber, S.; Schneider, G.; Schäfer, H.; Arlt, A. TRAIL/NF-κB/CX3CL1 mediated onco-immuno crosstalk leading to TRAIL resistance of pancreatic cancer cell lines. Int. J. Mol. Sci. 2018, 19, 1661. [Google Scholar] [CrossRef] [PubMed]
- Geismann, C.; Grohmann, F.; Dreher, A.; Häsler, R.; Rosenstiel, P.; Legler, K.; Hauser, C.; Egberts, J.H.; Sipos, B.; Schreiber, S.; et al. Role of CCL20 mediated immune cell recruitment in NF-κB mediated TRAIL resistance of pancreatic cancer. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 782–796. [Google Scholar] [CrossRef] [PubMed]
- Riedlinger, T.; Haas, J.; Busch, J.; van de Sluis, B.; Kracht, M.; Schmitz, M.L. The direct and indirect roles of NF-κB in cancer: Lessons from oncogenic fusion proteins and knock-in mice. Biomedicines 2018, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Awasthee, N.; Rai, V.; Chava, S.; Nallasamy, P.; Kunnumakkara, A.B.; Bishayee, A.; Chauhan, S.C.; Challagundla, K.B.; Gupta, S.C. Targeting IκB kinases for cancer therapy. Semin. Cancer Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Colomer, C.; Marruecos, L.; Vert, A.; Bigas, A.; Espinosa, L. NF-κB-independent roles in cancer. Biomedicines 2017, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, S.; Minassian, A.; Li, J.; Feng, P. Recent advances on viral manipulation of NF-κB signaling pathway. Curr. Opin. Virol. 2015, 15, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, A.S. Regulation of cell death and autophagy by IKK and NF-κB: Critical mechanisms in immune function and cancer. Immunol. Rev. 2012, 246, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor benefits of antiviral immunity: An underappreciated aspect of oncolytic virotherapies. Trends Immunol. 2018, 39, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Hoffner, B.; Gasal, E.; Hong, J.; Carvajal, R.D. Oncolytic immunotherapy: Unlocking the potential of viruses to help target cancer. Cancer Immunol. Immunother. 2017, 66, 1249–1264. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroun, J.; Muñoz-Alía, M.; Ammayappan, A.; Schulze, A.; Peng, K.-W.; Russell, S. Designing and building oncolytic viruses. Future Virol. 2017, 12, 193–213. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.T.; Bell, J.C. Oncolytic virus combination therapy: Killing one bird with two stones. Mol. Ther. 2018, 26, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Jhawar, S.R.; Thandoni, A.; Bommareddy, P.K.; Hassan, S.; Kohlhapp, F.J.; Goyal, S.; Schenkel, J.M.; Silk, A.W.; Zloza, A. Oncolytic viruses-natural and genetically engineered cancer immunotherapies. Front. Oncol. 2017, 7, 202. [Google Scholar] [CrossRef] [PubMed]
- Meyers, D.E.; Wang, A.A.; Thirukkumaran, C.M.; Morris, D.G. Current immunotherapeutic strategies to enhance oncolytic virotherapy. Front. Oncol. 2017, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.A.G. Converting cold into hot tumors by combining immunotherapies. Cell 2017, 170, 1055–1056. [Google Scholar] [CrossRef] [PubMed]
- Badalamenti, G.; Fanale, D.; Incorvaia, L.; Barraco, N.; Listì, A.; Maragliano, R.; Vincenzi, B.; Calò, V.; Iovanna, J.L.; Bazan, V.; et al. Role of tumor-infiltrating lymphocytes in patients with solid tumors: Can a drop dig a stone? Cell. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R.; Schatton, T.; Barthel, S.R. T-lymphocyte homing: An underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Investig. 2017, 97, 669–697. [Google Scholar] [CrossRef] [PubMed]
- Fournier, C.; Martin, F.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; Apetoh, L. Trial Watch: Adoptively transferred cells for anticancer immunotherapy. Oncoimmunology 2017, 6, e1363139. [Google Scholar] [CrossRef] [PubMed]
- Met, Ö.; Jensen, K.M.; Chamberlain, C.A.; Donia, M.; Svane, I.M. Principles of adoptive T cell therapy in cancer. Semin. Immunopathol. 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, Z.; Muranski, P.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 2013, 20, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.; Curti, B.D.; Hallmeyer, S.; Feng, Z.; Paustian, C.; Bifulco, C.; Fox, B.; Grose, M.; Shafren, D. Phase II calm extension study: Coxsackievirus A21 delivered intratumorally to patients with advanced melanoma induces immune-cell infiltration in the tumor microenvironment. J. Immunother. Cancer 2015, 3 (Suppl. 2), P343. [Google Scholar] [CrossRef]
- Alemany, R. Oncolytic adenoviruses in cancer treatment. Biomedicines 2014, 2, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Nagasato, M.; Yoshida, T.; Aoki, K. Recent advances in genetic modification of adenovirus vectors for cancer treatment. Cancer Sci. 2017, 108, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Liang, M. Oncorine, the world first oncolytic virus medicine and its update in China. Curr. Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Debbas, M.; White, E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993, 7, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Pilder, S.; Moore, M.; Logan, J.; Shenk, T. The adenovirus E1B-55K transforming polypeptide modulates transport or cytoplasmic stabilization of viral and host cell mRNAs. Mol. Cell. Biol. 1986, 6, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.T.; Aguirre-Hernández, C.; Halldén, G.; Parker, A.L. Designer oncolytic adenovirus: Coming of age. Cancers 2018, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- De Munck, J.; Binks, A.; McNeish, I.A.; Aerts, J.L. Oncolytic virus-induced cell death and immunity: A match made in heaven? J. Leukoc. Biol. 2017, 102, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Møller, A.W.; Garofalo, M.; Cerullo, V.; Pesonen, S.; Alemany, R.; Jaderberg, M. Antitumor-specific T-cell responses induced by oncolytic adenovirus ONCOS-102 (AdV5/3-D24-GM-CSF) in peritoneal mesothelioma mouse model. J. Med. Virol. 2018, 90, 1669–1673. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.; Ranki, T.; Joensuu, T.; Jäger, E.; Karbach, J.; Wahle, C.; Partanen, K.; Kairemo, K.; Alanko, T.; Turkki, R.; et al. Repeated intratumoral administration of ONCOS-102 leads to systemic antitumor CD8+ T-cell response and robust cellular and transcriptional immune activation at tumor site in a patient with ovarian cancer. Oncoimmunology 2015, 4, e1017702. [Google Scholar] [CrossRef] [PubMed]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimäki, A.; et al. Phase I study with ONCOS-102 for the treatment of solid tumors—An evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Abei, M.; Ugai, H.; Seo, E.; Wakayama, M.; Murata, T.; Todoroki, T.; Tanaka, N.; Hamada, H.; Yokoyama, K.K. E1A, E1B double-restricted adenovirus for oncolytic gene therapy of gallbladder cancer. Cancer Res. 2003, 63, 4434–4440. [Google Scholar] [PubMed]
- Yamada, K.; Moriyama, H.; Yasuda, H.; Hara, K.; Maniwa, Y.; Hamada, H.; Yokono, K.; Nagata, M. Modification of the Rb-binding domain of replication-competent adenoviral vector enhances cytotoxicity against human esophageal cancers via NF-κB activity. Hum. Gene Ther. 2007, 18, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Yamano, S.; Tokino, T.; Yasuda, M.; Kaneuchi, M.; Takahashi, M.; Niitsu, Y.; Fujinaga, K.; Yamashita, T. Induction of transformation and p53-dependent apoptosis by adenovirus type 5 E4orf6/7 cDNA. J. Virol. 1999, 73, 10095–10103. [Google Scholar] [PubMed]
- Passaro, C.; Borriello, F.; Vastolo, V.; Di Somma, S.; Scamardella, E.; Gigantino, V.; Franco, R.; Marone, G.; Portella, G. The oncolytic virus dl922-947 reduces IL-8/CXCL8 and MCP-1/CCL2 expression and impairs angiogenesis and macrophage infiltration in anaplastic thyroid carcinoma. Oncotarget 2016, 7, 1500–1515. [Google Scholar] [CrossRef] [PubMed]
- Wennier, S.; Li, S.; McFadden, G. Oncolytic virotherapy for pancreatic cancer. Expert Rev. Mol. Med. 2011, 13, e18. [Google Scholar] [CrossRef] [PubMed]
- Maliandi, M.V.; Mato-Berciano, A.; Sobrevals, L.; Roué, G.; José, A.; Fillat, C. AduPARE1A and gemcitabine combined treatment trigger synergistic antitumor effects in pancreatic cancer through NF-κB mediated uPAR activation. Mol. Cancer 2015, 14, 146. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.-H.; Wechman, S.L.; McMasters, K.M.; Zhou, H.S. Oncolytic replication of E1b-deleted adenoviruses. Viruses 2015, 7, 5767–5779. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.H.; Chen, M.-J.; Searle, P.F.; Kerr, D.J.; Young, L.S. Inhibition of NF-κB enhances the cytotoxicity of virus-directed enzyme prodrug therapy and oncolytic adenovirus cancer gene therapy. Gene Ther. 2005, 12, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaack, J.; Bennett, M.L.; Colbert, J.D.; Torres, A.V.; Clayton, G.H.; Ornelles, D.; Moorhead, J. E1A and E1B proteins inhibit inflammation induced by adenovirus. Proc. Natl. Acad. Sci. USA 2004, 101, 3124–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radke, J.R.; Grigera, F.; Ucker, D.S.; Cook, J.L. Adenovirus E1B 19-kilodalton protein modulates innate immunity through apoptotic mimicry. J. Virol. 2014, 88, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- Cerullo, V.; Diaconu, I.; Romano, V.; Hirvinen, M.; Ugolini, M.; Escutenaire, S.; Holm, S.-L.; Kipar, A.; Kanerva, A.; Hemminki, A. An oncolytic adenovirus enhanced for Toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol. Ther. 2012, 20, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Hirvinen, M.; Garofalo, M.; Romaniuk, D.; Kuryk, L.; Sarvela, T.; Vitale, A.; Antopolsky, M.; Magarkar, A.; Viitala, T.; et al. Oncolytic adenoviruses coated with MHC-I tumor epitopes increase the antitumor immunity and efficacy against melanoma. Oncoimmunology 2015, 5, e1105429. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Iovine, B.; Kuryk, L.; Capasso, C.; Hirvinen, M.; Vitale, A.; Yliperttula, M.; Bevilacqua, M.A.; Cerullo, V. Oncolytic adenovirus loaded with L-carnosine as novel strategy to enhance the antitumor activity. Mol. Cancer Ther. 2016, 15, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Saari, H.; Somersalo, P.; Crescenti, D.; Kuryk, L.; Aksela, L.; Capasso, C.; Madetoja, M.; Koskinen, K.; Oksanen, T.; et al. Antitumor effect of oncolytic virus and paclitaxel encapsulated in extracellular vesicles for lung cancer treatment. J. Control. Release 2018, 283, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Sanchala, D.S.; Bhatt, L.K.; Prabhavalkar, K.S. Oncolytic herpes simplex viral therapy: A stride toward selective targeting of cancer cells. Front. Pharmacol. 2017, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, N.A.S.; Rizos, H.; Diefenbach, R.J. Oncolytic virotherapy using herpes simplex virus: How far have we come? Oncolytic Virother. 2015, 4, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Fountzilas, C.; Patel, S.; Mahalingam, D. Review: Oncolytic virotherapy, updates and future directions. Oncotarget 2017, 8, 102617–102639. [Google Scholar] [CrossRef] [PubMed]
- Kulu, Y.; Kawasaki, H.; Donahue, J.M.; Kasuya, H.; Cusack, J.C.; Choi, E.W.; Kuruppu, D.K.; Fuchs, B.C.; Tanabe, K.K. Concurrent chemotherapy inhibits herpes simplex virus-1 replication and oncolysis. Cancer Gene Ther. 2013, 20, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsura, T.; Iwai, S.; Ota, Y.; Shimizu, H.; Ikuta, K.; Yura, Y. The effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cells. Cancer Gene Ther. 2009, 16, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.; Dicker, A.; Popa, C.; Jones, S.; Dahler, A. Histone deacetylase inhibitors as potential anti-skin cancer agents. Cancer Res. 1999, 59, 399–404. [Google Scholar] [PubMed]
- Minucci, S.; Horn, V.; Bhattacharyya, N.; Russanova, V.; Ogryzko, V.V.; Gabriele, L.; Howard, B.H.; Ozato, K. A histone deacetylase inhibitor potentiates retinoid receptor action in embryonal carcinoma cells. Proc. Natl. Acad. Sci. USA 1997, 94, 11295–11300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreansky, S.; Soroceanu, L.; Flotte, E.R.; Chou, J.; Markert, J.M.; Gillespie, G.Y.; Roizman, B.; Whitley, R.J. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res. 1997, 57, 1502–1509. [Google Scholar] [PubMed]
- Antoszczyk, S.; Spyra, M.; Mautner, V.F.; Kurtz, A.; Stemmer-Rachamimov, A.O.; Martuza, R.L.; Rabkin, S.D. Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus. Neuro Oncol. 2014, 16, 1057–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, J.D.; Markert, J.M.; Li, L.; Carroll, S.L.; Cassady, K.A. STAT1 and NF-κB inhibitors diminish basal interferon-stimulated gene expression and improve the productive infection of oncolytic HSV in MPNST cells. Mol. Cancer Res. 2016, 14, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Rezvani, K.; Shaim, H.; Hofs, E.; Ngom, M.; Bu, L.; Liu, G.; Lee, J.T.C.; Imren, S.; Lam, V.; et al. UV-inactivated HSV-1 potently activates NK cell killing of leukemic cells. Blood 2016, 127, 2575–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, A.L.; Barf, M.; Geletneky, K.; Unterberg, A.; Rommelaere, J. Immunotherapeutic potential of oncolytic H-1 parvovirus: Hints of glioblastoma microenvironment conversion towards immunogenicity. Viruses 2017, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.H.; Zeidler, M.; Wilsberg, V.; Cornelis, J.J.; Woelfel, T.; Rommelaere, J.; Galle, P.R.; Heike, M. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum. Gene Ther. 2005, 16, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Sieben, M.; Schäfer, P.; Dinsart, C.; Galle, P.R.; Moehler, M. Activation of the human immune system via Toll-like receptors by the oncolytic parvovirus H-1. Int. J. Cancer 2013, 132, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Rouanet, M.; Lebrin, M.; Gross, F.; Bournet, B.; Cordelier, P.; Buscail, L. Gene therapy for pancreatic cancer: Specificity, issues and hopes. Int. J. Mol. Sci. 2017, 18, 1231. [Google Scholar] [CrossRef] [PubMed]
- Réjiba, S.; Bigand, C.; Parmentier, C.; Masmoudi, A.; Hajri, A. Oncosuppressive suicide gene virotherapy “PVH1-yCD/5-FC” for pancreatic peritoneal carcinomatosis treatment: NFκB and Akt/PI3K involvement. PLoS ONE 2013, 8, e70594. [Google Scholar] [CrossRef] [PubMed]
- Carocci, M.; Bakkali-Kassimi, L. The encephalomyocarditis virus. Virulence 2012, 3, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, F.C.; Roberts, A.M.; Hwang, I.I.L.; Moriyama, E.H.; Evans, A.J.; Sybingco, S.; Watson, I.R.; Carneiro, L.A.M.; Gedye, C.; Girardin, S.E.; et al. Oncolytic targeting of renal cell carcinoma via encephalomyocarditis virus. EMBO Mol. Med. 2010, 2, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-M.; Kim, M.-H.; Kim, B.H.; Jung, S.-H.; Kim, Y.S.; Park, H.J.; Hong, J.T.; Min, K.R.; Kim, Y. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-κB without affecting IκB degradation. FEBS Lett. 2004, 571, 50–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, X.; Tan, J.; Lim, K.J.; Koh, J.; Ooi, W.F.; Li, Z.; Huang, D.; Xing, M.; Chan, Y.S.; Qu, J.Z.; et al. VHL deficiency drives enhancer activation of oncogenes in clear cell renal cell carcinoma. Cancer Discov. 2017, 7, 1284–1305. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Pattoli, M.A.; Gregor, K.R.; Brassil, P.J.; MacMaster, J.F.; McIntyre, K.W.; Yang, X.; Iotzova, V.S.; Clarke, W.; Strnad, J.; et al. BMS-345541 is a highly selective inhibitor of IκB kinase that binds at an allosteric site of the enzyme and blocks NF-κB-dependent transcription in mice. J. Biol. Chem. 2003, 278, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Podolin, P.L.; Callahan, J.F.; Bolognese, B.J.; Li, Y.H.; Carlson, K.; Davis, T.G.; Mellow, G.W.; Evans, C.; Roshak, A.K. Attenuation of murine collagen-induced arthritis by a novel, potent, selective small molecule inhibitor of IκB kinase 2, TPCA-1 (2-[(Aminocarbonyl)amino]-5-(4-fluorophenyl)-3-thiophenecarboxamide), occurs via reduction of proinflammatory cytokines and antigen-induced T cell proliferation. J. Pharmacol. Exp. Ther. 2005, 312, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Whitt, M.A.; Baumann, J.; Garner, J.M.; Morton, C.L.; Davidoff, A.M.; Pfeffer, L.M. Inhibition of type I interferon-mediated antiviral action in human glioma cells by the IKK inhibitors BMS-345541 and TPCA-1. J. Interferon Cytokine Res. 2012, 32, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Wang, B.-R.; Wu, Y.-Q.; Wang, F.-C.; Zhang, J.; Wang, Y.-G. Oncolytic viruses against cancer stem cells: A promising approach for gastrointestinal cancer. World J. Gastroenterol. 2016, 22, 7999–8009. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Oncolytic alphaviruses in cancer immunotherapy. Vaccines 2017, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. New frontiers in oncolytic viruses: Optimizing and selecting for virus strains with improved efficacy. Biologics 2018, 12, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Chijiwa, T.; Mishima, A.; Hagiwara, M.; Sano, M.; Hayashi, K.; Inoue, T.; Naito, K.; Toshioka, T.; Hidaka, H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H89), of PC12D pheochromocytoma cells. J. Biol. Chem. 1990, 265, 5267–5272. [Google Scholar] [PubMed]
- Muroi, M.; Suzuki, T. Role of protein kinase A in LPS-induced activation of NF-κB proteins of a mouse macrophage-like cell line, J774. Cell. Signal. 1993, 5, 289–298. [Google Scholar] [CrossRef]
- Koga, K.; Takaesu, G.; Yoshida, R.; Nakaya, M.; Kobayashi, T.; Kinjyo, I.; Yoshimura, A. Cyclic adenosine monophosphate suppresses the transcription of proinflammatory cytokines via the phosphorylated c-Fos protein. Immunity 2009, 30, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liang, J.; Lin, Y.; Zhang, H.; Xiao, X.; Tan, Y.; Cai, J.; Zhu, W.; Xing, F.; Hu, J.; et al. A classical PKA inhibitor increases the oncolytic effect of M1 virus via activation of exchange protein directly activated by cAMP 1. Oncotarget 2016, 7, 48443–48455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyanasundram, J.; Hamid, A.; Yusoff, K.; Chia, S.L. Newcastle disease virus strain AF2240 as an oncolytic virus: A review. Acta Trop. 2018, 183, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Ch’ng, W.-C.; Abd-Aziz, N.; Ong, M.-H.; Stanbridge, E.J.; Shafee, N. Human renal carcinoma cells respond to Newcastle disease virus infection through activation of the p38 MAPK/NF-κB/IκBα pathway. Cell. Oncol. 2015, 38, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.; Morris, D.G. Oncolytic viral therapy using reovirus. Methods Mol. Biol. 2015, 1317, 187–223. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.L.; Rodgers, S.E.; Clarke, P.; Ballard, D.W.; Kerr, L.D.; Tyler, K.L.; Dermody, T.S. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J. Virol. 2000, 74, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.; Meintzer, S.M.; Moffitt, L.A.; Tyler, K.L. Two distinct phases of virus-induced nuclear factor κB regulation enhance tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in virus-infected cells. J. Biol. Chem. 2003, 278, 18092–18100. [Google Scholar] [CrossRef] [PubMed]
- Min, H.-J.; Koh, S.S.; Cho, I.-R.; Srisuttee, R.; Park, E.-H.; Jhun, B.H.; Kim, Y.-G.; Oh, S.; Kwak, J.E.; Johnston, R.N.; et al. Inhibition of GSK-3β enhances reovirus-induced apoptosis in colon cancer cells. Int. J. Oncol. 2009, 35, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Ougolkov, A.V.; Fernandez-Zapico, M.E.; Savoy, D.N.; Urrutia, R.A.; Billadeau, D.D. Glycogen synthase kinase-3β participates in nuclear factor κB–mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005, 65, 2076–2081. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormö, M.; Nilsson, Y.; Radesäter, A.-C.; Jerning, E.; Markgren, P.-O.; Borgegård, T.; et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, D.; Tovar, C.; Yang, H.; Vu, B.T.; Heimbrook, D.C.; Vassilev, L.T. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res. 2005, 65, 1918–1924. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Pan, L.-Z.; Hill, R.; Marcato, P.; Shmulevitz, M.; Vassilev, L.T.; Lee, P.W.K. Stabilisation of p53 enhances reovirus-induced apoptosis and virus spread through p53-dependent NF-κB activation. Br. J. Cancer 2011, 105, 1012–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, R.; Mookerjee, B.; van Hensbergen, Y.; Bedi, G.C.; Giordano, A.; El-Deiry, W.S.; Fuchs, E.J.; Bedi, A. p53-mediated repression of nuclear factor-κB RelA via the transcriptional integrator p300. Cancer Res. 1998, 58, 4531–4536. [Google Scholar] [PubMed]
- Wu, H.; Lozano, G. NF-κB activation of p53. A potential mechanism for suppressing cell growth in response to stress. J. Biol. Chem. 1994, 269, 20067–20074. [Google Scholar] [PubMed]
- Pan, D.; Marcato, P.; Ahn, D.-G.; Gujar, S.; Pan, L.-Z.; Shmulevitz, M.; Lee, P.W.K. Activation of p53 by chemotherapeutic agents enhances reovirus oncolysis. PLoS ONE 2013, 8, e54006. [Google Scholar] [CrossRef] [PubMed]
- Basile, J.R.; Eichten, A.; Zacny, V.; Münger, K. NF-κB-mediated induction of p21Cip1/Waf1 by tumor necrosis factor α induces growth arrest and cytoprotection in normal human keratinocytes. Mol. Cancer Res. 2003, 1, 262–270. [Google Scholar] [PubMed]
- Park, Y.H. The nuclear factor-κB pathway and response to treatment in breast cancer. Pharmacogenomics 2017, 18, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Thirukkumaran, C.; Shi, Z.-Q.; Thirukkumaran, P.; Luider, J.; Kopciuk, K.; Spurrell, J.; Elzinga, K.; Morris, D. PUMA and NF-κB are cell signaling predictors of reovirus oncolysis of breast cancer. PLoS ONE 2017, 12, e0168233. [Google Scholar] [CrossRef] [PubMed]
- Salimi, V.; Tavakoli-Yaraki, M.; Mahmoodi, M.; Shahabi, S.; Gharagozlou, M.J.; Shokri, F.; Mokhtari-Azad, T. The oncolytic effect of respiratory syncytial virus (RSV) in human skin cancer cell line, A431. Iran. Red Crescent Med. J. 2013, 15, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Echchgadda, I.; Kota, S.; DeLa Cruz, I.; Sabbah, A.; Chang, T.; Harnack, R.; Mgbemena, V.; Chatterjee, B.; Bose, S. Anticancer oncolytic activity of respiratory syncytial virus. Cancer Gene Ther. 2009, 16, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Echchgadda, I.; Chang, T.-H.; Sabbah, A.; Bakri, I.; Ikeno, Y.; Hubbard, G.B.; Chatterjee, B.; Bose, S. Oncolytic targeting of androgen-sensitive prostate tumor by the respiratory syncytial virus (RSV): Consequences of deficient interferon-dependent antiviral defense. BMC Cancer 2011, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Felt, S.A.; Grdzelishvili, V.Z. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: A 5-year update. J. Gen. Virol. 2017, 98, 2895–2911. [Google Scholar] [CrossRef] [PubMed]
- Shulak, L.; Beljanski, V.; Chiang, C.; Dutta, S.M.; Van Grevenynghe, J.; Belgnaoui, S.M.; Nguyên, T.L.-A.; Di Lenardo, T.; Semmes, O.J.; Lin, R.; et al. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-κB-dependent autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Denlinger, C.E.; Rundall, B.K.; Smith, P.W.; Jones, D.R. Suberoylanilide hydroxamic acid induces Akt-mediated phosphorylation of p300, which promotes acetylation and transcriptional activation of RelA/p65. J. Biol. Chem. 2006, 281, 31359–31368. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.M.-Y. Inhibition of tumor necrosis factor by curcumin, a phytochemical. Biochem. Pharmacol. 1995, 49, 1551–1556. [Google Scholar] [CrossRef]
- Fehl, D.J.; Ahmed, M. Curcumin promotes the oncolytic capacity of vesicular stomatitis virus for the treatment of prostate cancers. Virus Res. 2017, 228, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Behnke, M.; Chen, S.; Cruickshank, A.A.; Dick, L.R.; Grenier, L.; Klunder, J.M.; Ma, Y.-T.; Plamondon, L.; Stein, R.L. Potent and selective inhibitors of the proteasome: Dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998, 8, 333–338. [Google Scholar] [CrossRef]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef]
- Yarde, D.N.; Nace, R.A.; Russell, S.J. Oncolytic vesicular stomatitis virus and bortezomib are antagonistic against myeloma cells in vitro but have additive anti-myeloma activity in vivo. Exp. Hematol. 2013, 41, 1038–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, S.; Nace, R.; Federspiel, M.J.; Barber, G.N.; Peng, K.-W.; Russell, S.J. Curative one-shot systemic virotherapy in murine myeloma. Leukemia 2012, 26, 1870–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| OV | Treatment | Condition | Status | Phase | References |
|---|---|---|---|---|---|
| AV | Ad5-yCD/mutTKSR39rep-ADP (Theragene®) | Non-small cell lung cancer stage I | Recruiting | I | NCT03029871 |
| 5-FC | |||||
| vGCV | |||||
| SBRT | |||||
| Ad5-yCD/mutTKSR39rep-hIL12 | Prostate cancer | Recruiting | I | NCT02555397 | |
| Ad5-yCD/mutTKSR39rep-hIL12 | Metastatic pancreatic cancer | Recruiting | I | NCT03281382 | |
| 5-FC | |||||
| ADV/HSV-tk | Metastatic non-small cell lung cancer Metastatic triple-negative breast cancer | Recruiting | II | NCT03004183 | |
| Valacyclovir | |||||
| SBRT | |||||
| CG0070 | Bladder cancer | Active, not recruiting | II | NCT02365818 | |
| DNX-2401 (Delta-24-RGD-4C) | Brainstem glioma | Recruiting | I | NCT03178032 | |
| Neoadjuvant therapy | |||||
| DNX-2401 (Delta-24-RGD-4C) | Glioblastoma | Recruiting | II | NCT02798406 | |
| Pembrolizumab | Gliosarcoma | ||||
| DNX-2440 | Recurrent glioblastoma | Recruiting | I | NCT03714334 | |
| LOAd703 | Biliary carcinoma | Recruiting | I/II | NCT03225989 | |
| Colorectal cancer | |||||
| Ovarian cancer | |||||
| Pancreatic adenocarcinoma | |||||
| LOAd703 | Pancreatic cancer | Recruiting | I/II | NCT02705196 | |
| Gemcitabine | |||||
| Nab-paclitaxel | |||||
| NSC-CRAd-S-pk7 | Malignant glioma | Recruiting | I | NCT03072134 | |
| OBP-301 (Telomelysin) | Melanoma stage III-IV | Recruiting | II | NCT03190824 | |
| ONCOS-102 | Castration-resistant prostate cancer | Recruiting | I/II | NCT03514836 | |
| Cyclophosphamide | |||||
| DCVac/PCa | |||||
| ONCOS-102 | Advanced or unresectable melanoma progressing after PD-1 blockade | Recruiting | I | NCT03003676 | |
| Cyclophosphamide | |||||
| Pembrolizumab | |||||
| ONCOS-102 | Malignant pleural mesothelioma | Recruiting | I/II | NCT02879669 | |
| Cyclophosphamide | |||||
| Pemetrexed/cisplatin (carboplatin) | |||||
| VCN-01 | Locally advanced solid tumors | Recruiting | I | NCT02045602 | |
| Gemcitabine | Metastatic solid tumors | ||||
| Abraxane® | Pancreatic adenocarcinoma | ||||
| HSV-1 | C134 | Recurrent malignant glioma | Not yet recruiting | I | NCT03657576 |
| G207 | Supratentorial neoplasms, malignant | Recruiting | I | NCT02457845 | |
| M032 (NSC 733972) | Recurrent malignant glioma | Recruiting | I | NCT02062827 | |
| rQNestin34.5v.2 (rQNestin) | Recurrent malignant glioma | Recruiting | I | NCT03152318 | |
| Cyclophosphamide | |||||
| TBI-1401(HF10) (Canerpaturev) | Pancreatic cancer stage III-IV | Recruiting | I | NCT03252808 | |
| Gemcitabine | |||||
| Nab-paclitaxel | |||||
| TS-1 | |||||
| TBI-1401(HF10) (Canerpaturev) | Melanoma stage III-IV | Active, not recruiting | II | NCT03153085 | |
| Ipilimumab | |||||
| T-VEC | Basal cell carcinoma | Recruiting | I | NCT03458117 | |
| Cutaneous lymphoma | |||||
| Merkel cell carcinoma | |||||
| Non-melanoma skin cancer | |||||
| Squamous cell carcinoma | |||||
| Recurrent breast cancer that cannot be removed by surgery | Active, not recruiting | II | NCT02658812 | ||
| T-VEC | Metastatic, unresectable, or recurrent HER2- negative breast cancer | Not yet recruiting | I | NCT03554044 | |
| Anastrozole | |||||
| Exemestane | |||||
| Fulvestrant | |||||
| Letrozole | |||||
| Paclitaxel | |||||
| Tamoxifen | |||||
| T-VEC | Locally advanced or metastatic rectal cancer | Recruiting | I | NCT03300544 | |
| Capecitabine | |||||
| Fluorouracil | |||||
| Oxaliplatin | |||||
| T-VEC | Refractory T cell and NK cell lymphomas Cutaneous squamous cell carcinoma Merkel cell carcinoma Other rare skin tumors | Recruiting | II | NCT02978625 | |
| Nivolumab | |||||
| T-VEC Paclitaxel | Breast cancer Ductal carcinoma Invasive breast carcinoma Invasive ductal breast carcinoma | Recruiting | I/II | NCT02779855 | |
| T-VEC | Melanoma stage III-IV | Recruiting | II | NCT02965716 | |
| Pembrolizumab | |||||
| PV | H-1PV (ParvOryx™) | Metastatic inoperable pancreatic cancer | Recruiting | I/II | NCT02653313 |
| RV | REOLYSIN® | KRAS mutant metastatic colorectal cancer | Active, not recruiting | I | NCT01274624 |
| Irinotecan | |||||
| Leucovorin | |||||
| 5-FU | |||||
| Bevacizumab | |||||
| Wild-type Reovirus | Relapsed or refractory multiple myeloma | Active, not recruiting | I | NCT02514382 | |
| Bortezomib | |||||
| Dexamethasone | |||||
| Wild-type Reovirus | Recurrent plasma cell myeloma | Not yet recruiting | I | NCT03605719 | |
| Carfilzomib | |||||
| Dexamethasone | |||||
| Nivolumab | |||||
| Pomalidomide | |||||
| Wild-type Reovirus Paclitaxel | Recurrent fallopian tube carcinoma Recurrent ovarian carcinoma Recurrent primary peritoneal carcinoma | Active, not recruiting | II | NCT01199263 | |
| VSV | VSV-hIFNβ-NIS | Stage IV or recurrent endometrial cancer | Recruiting | I | NCT03120624 |
| Technetium Tc-99m Sodium Pertechnetate | |||||
| VSV-IFNβ-NIS | Malignant solid tumor | Recruiting | I/II | NCT02923466 | |
| VSV-IFNβ-NIS and Avelumab | |||||
| VSV-IFNβ-NIS | Refractory non-small cell lung cancer or Hepatocellular carcinoma | Not yet recruiting | I | NCT03647163 | |
| Pembrolizumab |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Struzik, J.; Szulc-Dąbrowska, L. NF-κB Signaling in Targeting Tumor Cells by Oncolytic Viruses—Therapeutic Perspectives. Cancers 2018, 10, 426. https://doi.org/10.3390/cancers10110426
Struzik J, Szulc-Dąbrowska L. NF-κB Signaling in Targeting Tumor Cells by Oncolytic Viruses—Therapeutic Perspectives. Cancers. 2018; 10(11):426. https://doi.org/10.3390/cancers10110426
Chicago/Turabian StyleStruzik, Justyna, and Lidia Szulc-Dąbrowska. 2018. "NF-κB Signaling in Targeting Tumor Cells by Oncolytic Viruses—Therapeutic Perspectives" Cancers 10, no. 11: 426. https://doi.org/10.3390/cancers10110426