Real-Time Monitoring and Detection of Single-Cell Level Cytokine Secretion Using LSPR Technology

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fabrication of Porous Alumina (PA) Mold
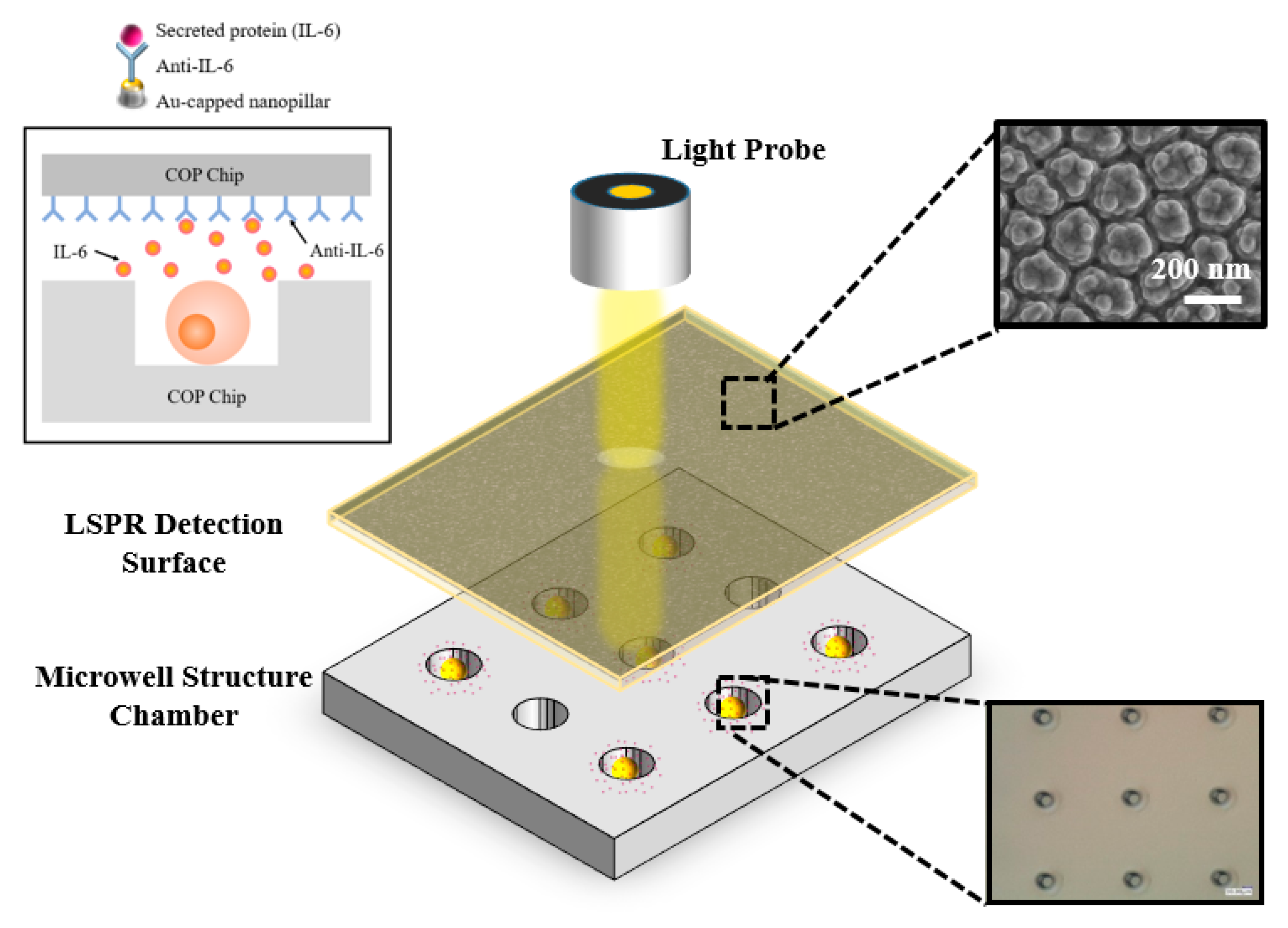
2.2. Fabrication and Immobilization of Gold-Capped Nanopillar Structured Polymer Film
2.3. Fabrication of Cell Trapping Micro-Well Structured Chip
2.4. Sensitivity Evaluation of the Gold-Capped COP Chip
2.5. Single Cell Occupancy Capability Evaluation of the Trapping Chip
2.6. Real-time Monitoring of Single Cell IL-6 Secretion Situation
3. Results and Discussion
3.1. Morphology Characterization of the COP Detection and Trapping Device
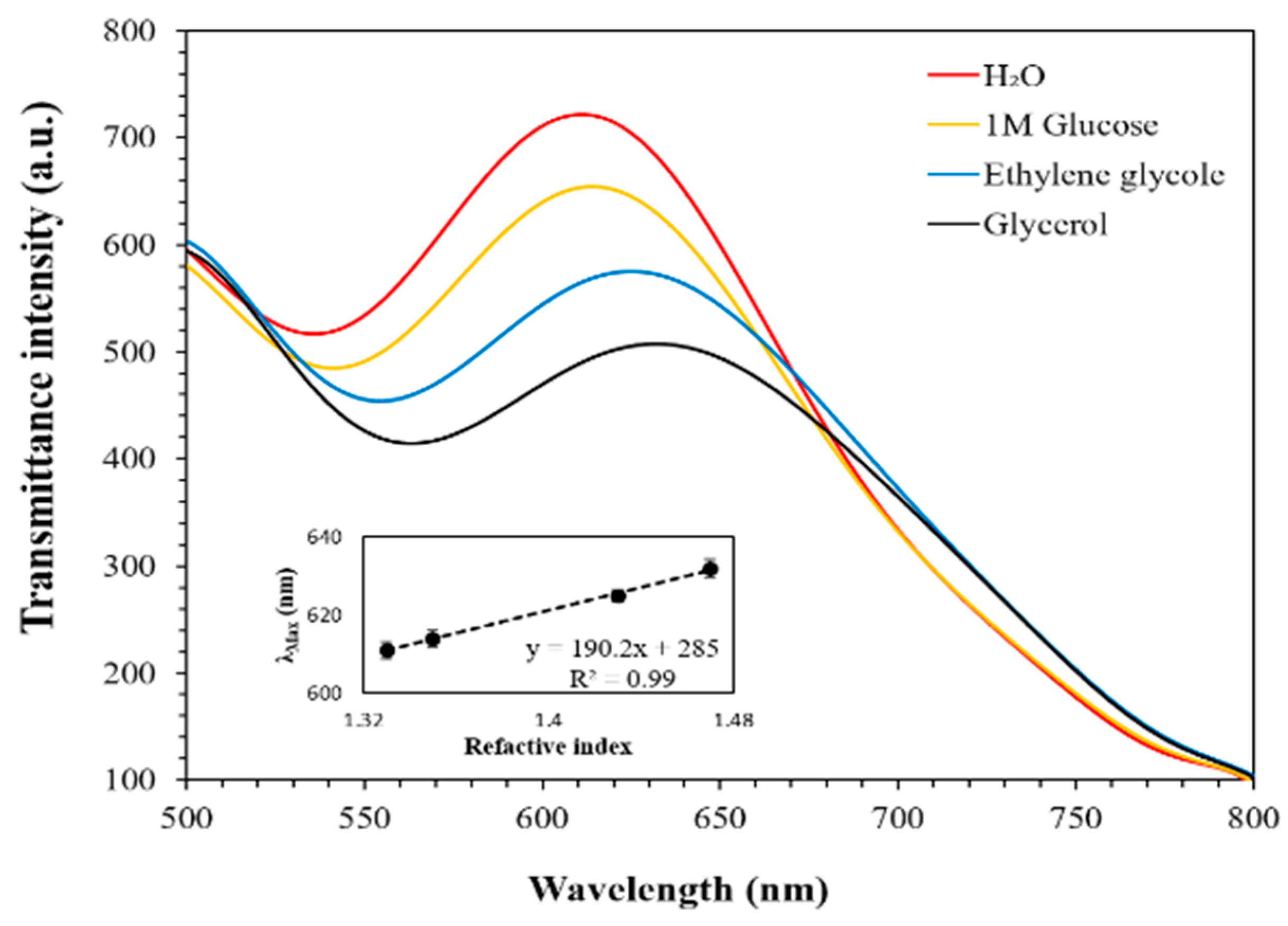
3.2. Sensitivity Evaluation of the COP Film via Transmittance Spectrum Peak Red-Shift
3.3. IL-6 Calibration Curve Detection Based on COP Detection Film
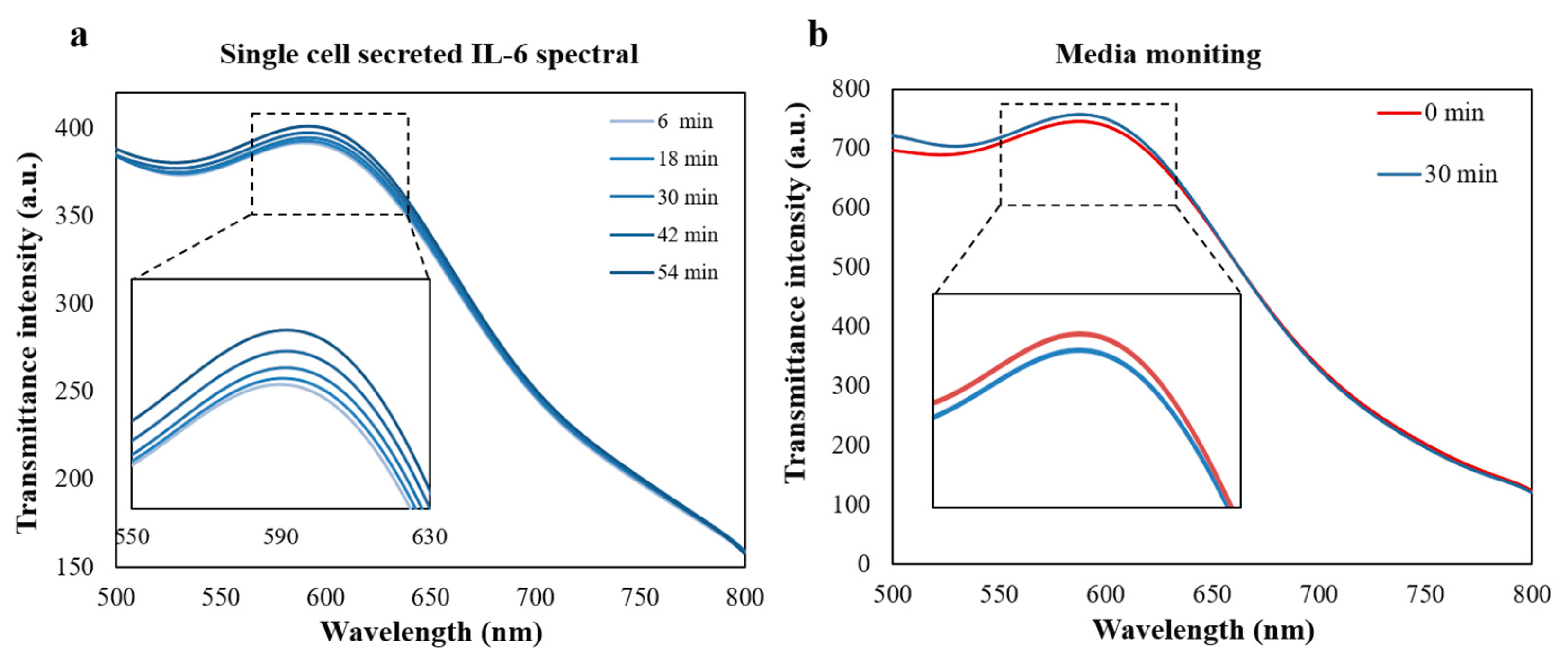
3.4. Single Cell Occupancy Performance and Real-Time Monitoring of Single Cell Cytokine Secretion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Benton, H.P. Cytokines and their receptors. Curr. Opin. Cell Biol. 1991, 3, 171–175. [Google Scholar] [CrossRef]
- Martins e Silva, J. [Biochemical characterization and metabolic effects of tumor necrosis factor]. Caracterizacao bioquimica e efeitos metabolicos do factor de necrose tumoral. Acta Med. Port. 1991, 4 (Suppl. 1), 20S–27S. [Google Scholar]
- Oh, B.R.; Huang, N.T.; Chen, W.Q.; Seo, J.H.; Chen, P.Y.; Cornell, T.T.; Shanley, T.P.; Fu, J.P.; Kurabayashi, K. Integrated nanoplasmonic sensing for cellular functional immunoanalysis using human blood. ACS Nano 2014, 8, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Kitamura, A.; Murahashi, M.; Yamanaka, K.; Hoa, L.Q.; Yamaguchi, Y.; Tamiya, E. Novel gold-capped nanopillars imprinted on a polymer film for highly sensitive plasmonic biosensing. Anal. Chem. 2012, 84, 5494–5500. [Google Scholar] [CrossRef] [PubMed]
- Fishman, D.; Faulds, G.; Jeffery, R.; Mohamed-Ali, V.; Yudkin, J.S.; Humphries, S.; Woo, P. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J. Clin. Investig. 1998, 102, 1369–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Hirano, T.; Taga, T.; Kishimoto, T. Biology of multifunctional cytokines—Il-6 and related molecules (Il-1 and Tnf). FASEB J. 1990, 4, 2860–2867. [Google Scholar] [CrossRef]
- Barton, B.E. IL-6: Insights into novel biological activities. Clin. Immunol. Immunopathol. 1997, 85, 16–20. [Google Scholar] [CrossRef]
- Schindler, R.; Mancilla, J.; Endres, S.; Ghorbani, R.; Clark, S.C.; Dinarello, C.A. Correlations and interactions in the production of Interleukin-6 (Il-6), Il-1, and tumor necrosis factor (Tnf) in human-blood mononuclear-cells—Il-6 suppresses Il-1 and Tnf. Blood 1990, 75, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Buss, H.; Chan, T.P.; Sluis, K.B.; Domigan, N.M.; Winterbourn, C.C. Protein carbonyl measurement by a sensitive ELISA method (vol 23, pg 361, 1997). Free Radic. Biol. Med. 1998, 24, 1352. [Google Scholar]
- Voller, A.; Bartlett, A.; Bidwell, D.E. Enzyme immunoassays with special reference to Elisa techniques. J. Clin. Pathol. 1978, 31, 507–520. [Google Scholar] [CrossRef] [Green Version]
- Lequin, R.M. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA). Clin. Chem. 2005, 51, 2415–2418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patiris, P.; Hanson, C. Single-round HIV type 1 neutralization measured by ELISPOT technique in primary human cells. AIDS Res. Hum. Retrovir. 2005, 21, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Fujihashi, K.; Mcghee, J.R.; Beagley, K.W.; Mcpherson, D.T.; Mcpherson, S.A.; Huang, C.M.; Kiyono, H. Cytokine-specific elispot assay—Single cell analysis of Il-2, Il-4 and Il-6 producing cells. J. Immunol. Methods 1993, 160, 181–189. [Google Scholar] [CrossRef]
- Okamoto, Y.; Gotoh, Y.; Tokui, H.; Mizuno, A.; Kobayashi, Y.; Nishida, M. Characterization of the cytokine network at a single cell level in mice with collagen-induced arthritis using a dual color ELISPOT assay. J. Interferon Cytokine Res. 2000, 20, 55–61. [Google Scholar] [CrossRef]
- Zand, M.S.; Henn, A.D.; Brown, M.A.; Rebhan, J.; Murphy, A.J.; Coca, M.N.; Hyrien, O.; Mosmann, T. Measurement of single-cell IgG secretion rates by quantitative ELISPOT (qELISPOT): Modulation by BCR-crosslinking and cell division in CpG stimulated IgG+CD27+ human memory B cells. Am. J. Transplant. 2009, 9, 471. [Google Scholar]
- Cohen, D.; Dickerson, J.A.; Whitmore, C.D.; Turner, E.H.; Palcic, M.M.; Hindsgaul, O.; Dovichi, N.J. Chemical cytometry: Fluorescence-based single-cell analysis. Annu. Rev. Anal. Chem. 2008, 1, 165–190. [Google Scholar] [CrossRef]
- Mark, H.F.L.; Rehan, J.; Mark, S.; Santoro, K.; Zolnierz, K. Fluorescence in situ hybridization analysis of single-cell trisomies for determination of clonality. Cancer Genet. Cytogenet. 1998, 102, 1–5. [Google Scholar] [CrossRef]
- Wang, Y.Z.; DelRosso, N.V.; Vaidyanathan, T.V.; Cahill, M.K.; Reitman, M.E.; Pittolo, S.; Mi, X.L.; Yu, G.Q.; Poskanzer, K.E. Accurate quantification of astrocyte and neurotransmitter fluorescence dynamics for single-cell and population-level physiology. Nat. Neurosci. 2019, 22, 1936–1944. [Google Scholar] [CrossRef]
- Hiep, H.M.; Nakayama, T.; Saito, M.; Yamamura, S.; Takamura, Y.; Tamiya, E. A microfluidic chip based on localized surface plasmon resonance for real-time monitoring of antigen-antibody reactions. Jpn. J. Appl. Phys. 2008, 47, 1337–1341. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Byun, J.Y.; Mun, H.; Shim, W.B.; Shin, Y.B.; Li, T.; Kim, M.G. A regeneratable, label-free, localized surface plasmon resonance (LSPR) aptasensor for the detection of ochratoxin A. Biosens. Bioelectron. 2014, 59, 321–327. [Google Scholar] [CrossRef]
- Raphael, M.P.; Christodoulides, J.A.; Delehanty, J.B.; Long, J.P.; Pehrsson, P.E.; Byers, J.M. Quantitative LSPR imaging for biosensing with single nanostructure resolution. Biophys. J. 2013, 104, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, B.; Angelome, P.C.; Lechuga, L.M.; Liz-Marzan, L.M. LSPR-based nanobiosensors. Nano Today 2009, 4, 244–251. [Google Scholar] [CrossRef]
- Bellapadrona, G.; Tesler, A.B.; Grunstein, D.; Hossain, L.H.; Kikkeri, R.; Seeberger, P.H.; Vaskevich, A.; Rubinstein, I. Optimization of localized surface plasmon resonance transducers for studying carbohydrate-protein interactions. Anal. Chem. 2012, 84, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Blaber, M.G.; Henry, A.I.; Bingham, J.M.; Schatz, G.C.; Van Duyne, R.P. LSPR imaging of silver triangular nanoprisms: Correlating scattering with structure using electrodynamics for plasmon lifetime analysis. J. Phys. Chem. C 2012, 116, 393–403. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Chen, Z.P.; Qu, C.L.; Chen, L.X. Highly sensitive visual detection of copper ions based on the shape-dependent LSPR spectroscopy of gold nanorods. Langmuir 2014, 30, 3625–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Nallathamby, P.D.; Xu, X.H.N. Photostable single-molecule nanoparticle optical biosensors for real-time sensing of single cytokine molecules and their binding reactions. J. Am. Chem. Soc. 2008, 130, 17095–17105. [Google Scholar] [CrossRef] [Green Version]
- Mayer, K.M.; Lee, S.; Liao, H.; Rostro, B.C.; Fuentes, A.; Scully, P.T.; Nehl, C.L.; Hafner, J.H. A label-free immunoassay based upon localized surface plasmon resonance of gold nanorods. ACS Nano 2008, 2, 687–692. [Google Scholar] [CrossRef]
- Wang, X.L.; Cui, Y.; Irudayaraj, J. Single-cell quantification of cytosine modifications by hyperspectral dark-field imaging. ACS Nano 2015, 9, 11924–11932. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, K.; Watarai, H.; Itoh, H.; Nakahama, E.; Ogawa, N. Measurement of antibody binding to protein immobilized on gold nanoparticles by localized surface plasmon spectroscopy. Anal. Bioanal. Chem. 2006, 386, 639–644. [Google Scholar] [CrossRef]
- Guo, L.H.; Kim, D.H. LSPR biomolecular assay with high sensitivity induced by aptamer-antigen-antibody sandwich complex. Biosens. Bioelectron. 2012, 31, 567–570. [Google Scholar] [CrossRef]
- Kim, D.K.; Park, T.J.; Tamiya, E.; Lee, S.Y. Label-free detection of leptin antibody-antigen interaction by using LSPR-based optical biosensor. J. Nanosci. Nanotechnol. 2011, 11, 4188–4193. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Hall, E.; Zare, R.N. Microfluidics-based cell culture for single-cell analysis. In Biophysical Journal; Biophysical Society: Rockville, MD, USA, 2007. [Google Scholar]
- Mazutis, L.; Gilbert, J.; Ung, W.L.; Weitz, D.A.; Griffiths, A.D.; Heyman, J.A. Single-cell analysis and sorting using droplet-based microfluidics. Nat. Protoc. 2013, 8, 870–891. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; Paguirigan, A.L.; Kreutz, J.E.; Radich, J.P.; Chiu, D.T. Microfluidics for single-cell genetic analysis. Lab Chip 2014, 14, 3135–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.B.; Marshall, D. Microfluidics for single cell analysis. Curr. Opin. Biotechnol. 2012, 23, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Andoh, A.; Hata, K.; Tasaki, K.; Araki, Y.; Fujiyama, Y.; Samba, T. IL-6 secretion by human pancreatic periacinar myofibroblasts in response to inflammatory mediators. J. Immunol. 2002, 168, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Vgontzas, A.N.; Bixler, E.O.; Lin, H.M.; Prolo, P.; Trakada, G.; Chrousos, G.P. IL-6 and its circadian secretion in humans. Neuroimmunomodulation 2005, 12, 131–140. [Google Scholar] [CrossRef]
- Hwang, S.J.; Tseng, M.C.; Shu, J.R.; Yu, H.H. Surface modification of cyclic olefin copolymer substrate by oxygen plasma treatment. Surf. Coat. Technol. 2008, 202, 3669–3674. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Occupancy Type | Single Cell | Double Cells | Multiple Cells |
|---|---|---|---|
| Occupancy Efficiency | ~60% | ~10% | ~5% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, C.; Luo, X.; Espulgar, W.V.; Koyama, S.; Kumanogoh, A.; Saito, M.; Takamatsu, H.; Tamiya, E. Real-Time Monitoring and Detection of Single-Cell Level Cytokine Secretion Using LSPR Technology. Micromachines 2020, 11, 107. https://doi.org/10.3390/mi11010107
Zhu C, Luo X, Espulgar WV, Koyama S, Kumanogoh A, Saito M, Takamatsu H, Tamiya E. Real-Time Monitoring and Detection of Single-Cell Level Cytokine Secretion Using LSPR Technology. Micromachines. 2020; 11(1):107. https://doi.org/10.3390/mi11010107
Chicago/Turabian StyleZhu, Chen, Xi Luo, Wilfred Villariza Espulgar, Shohei Koyama, Atsushi Kumanogoh, Masato Saito, Hyota Takamatsu, and Eiichi Tamiya. 2020. "Real-Time Monitoring and Detection of Single-Cell Level Cytokine Secretion Using LSPR Technology" Micromachines 11, no. 1: 107. https://doi.org/10.3390/mi11010107