Gelatin-Coated Microfluidic Channels for 3D Microtissue Formation: On-Chip Production and Characterization




Abstract
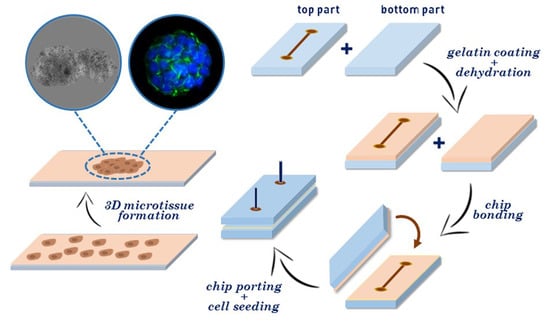
:
1. Introduction
2. Materials and Methods
2.1. Materials and Equipment
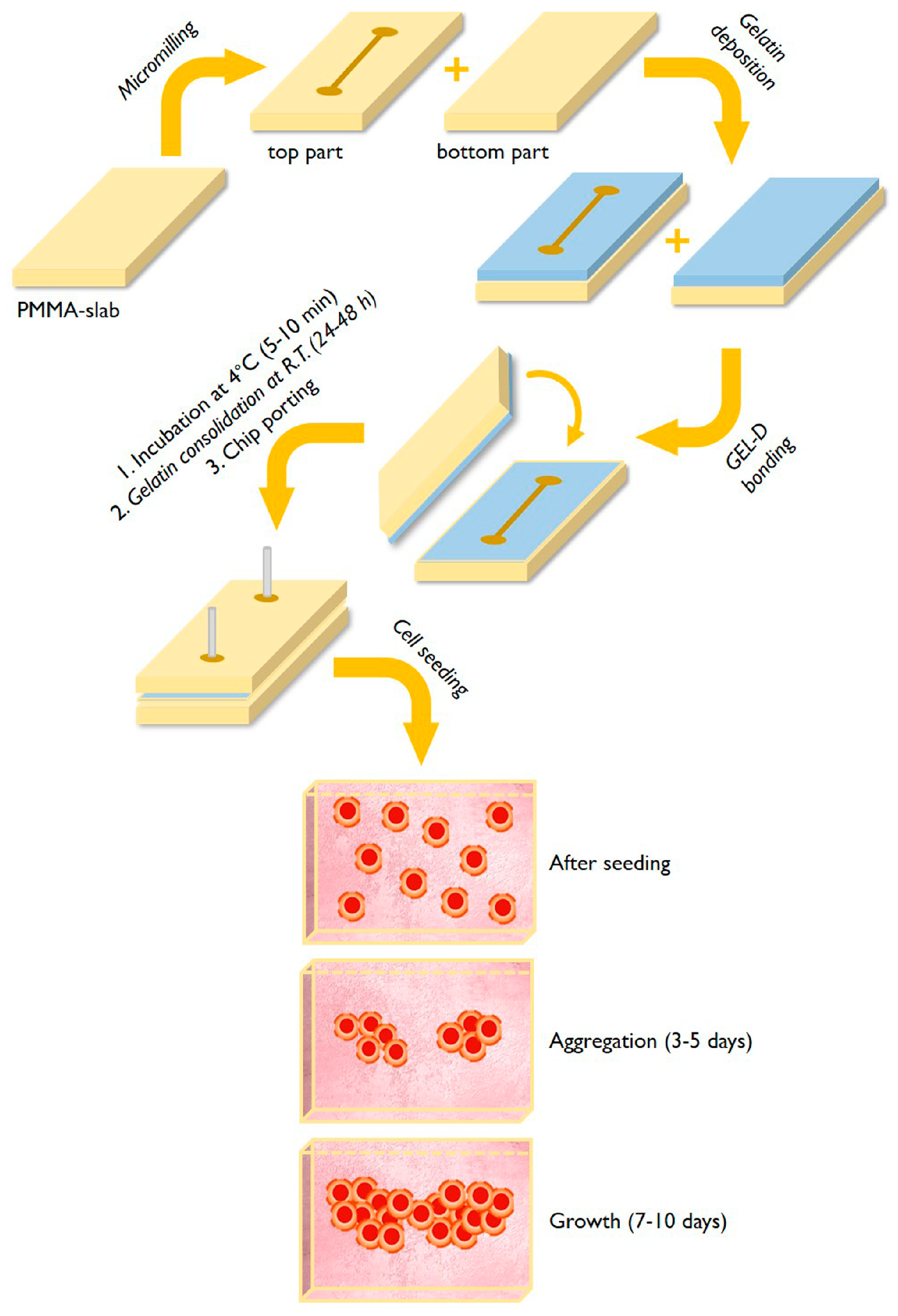
2.2. Fabrication of Microfluidic Chips and Gelatin Coating
2.3. Cell Seeding and Characterization
3. Results and Discussions
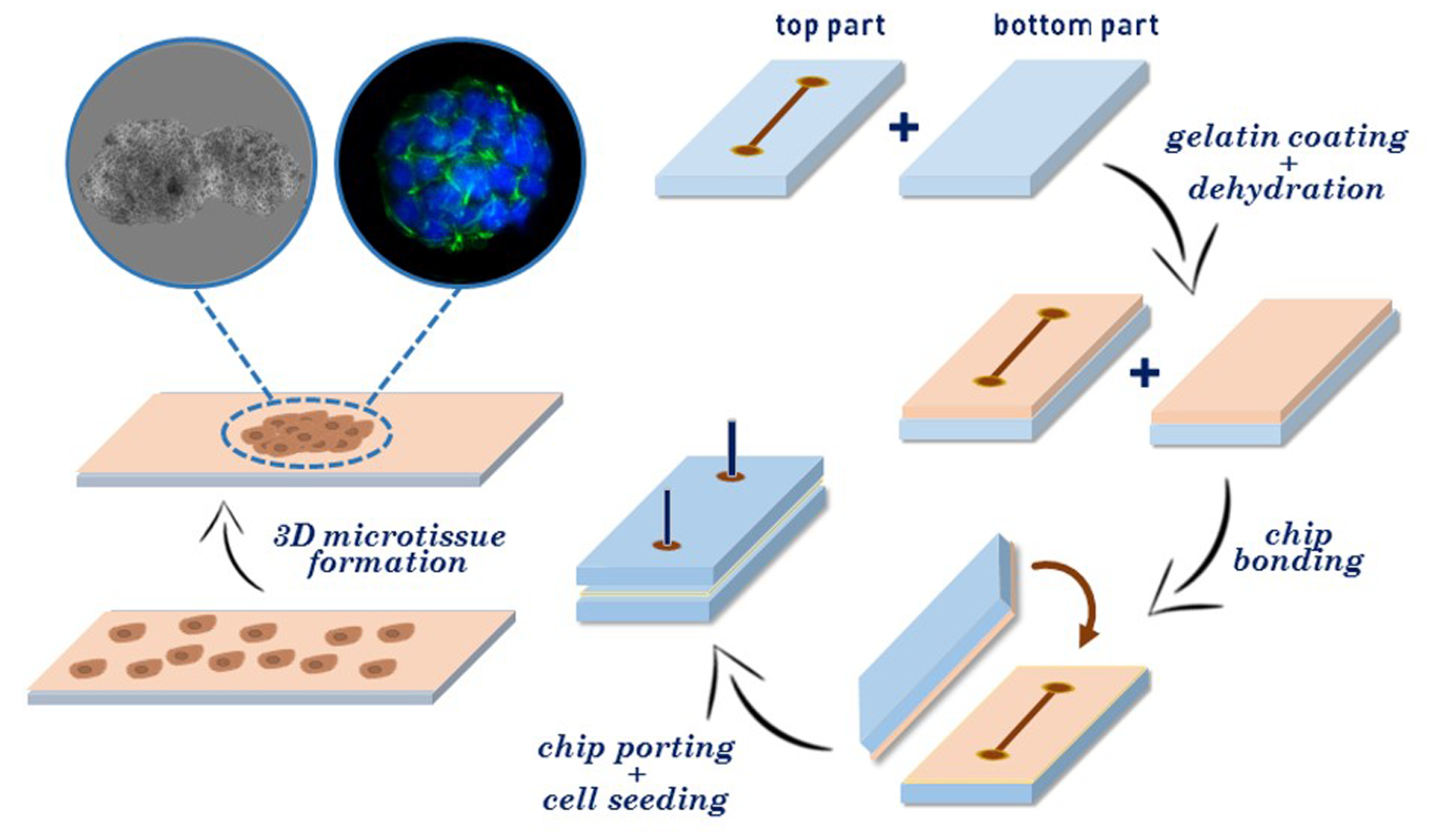
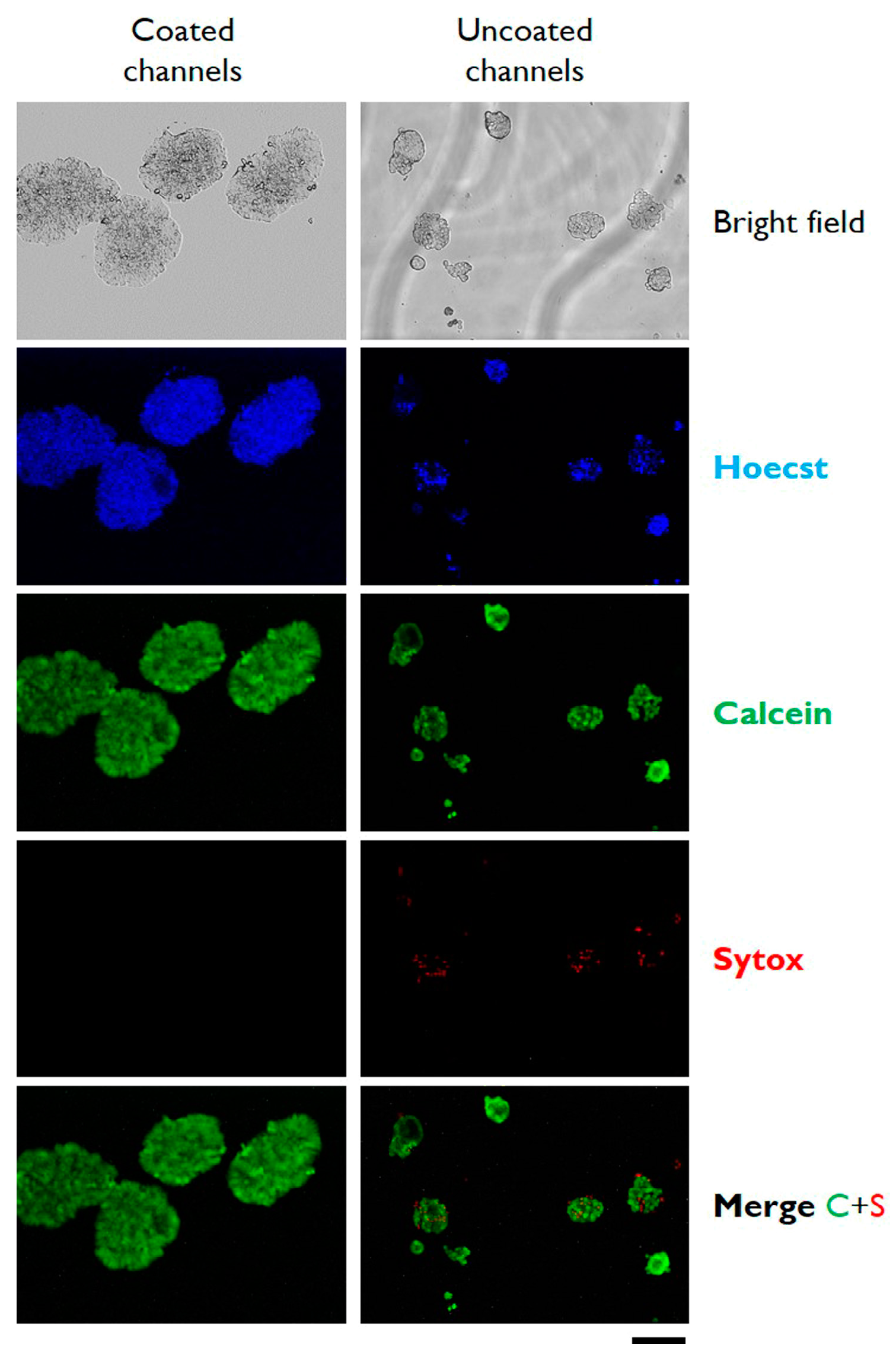
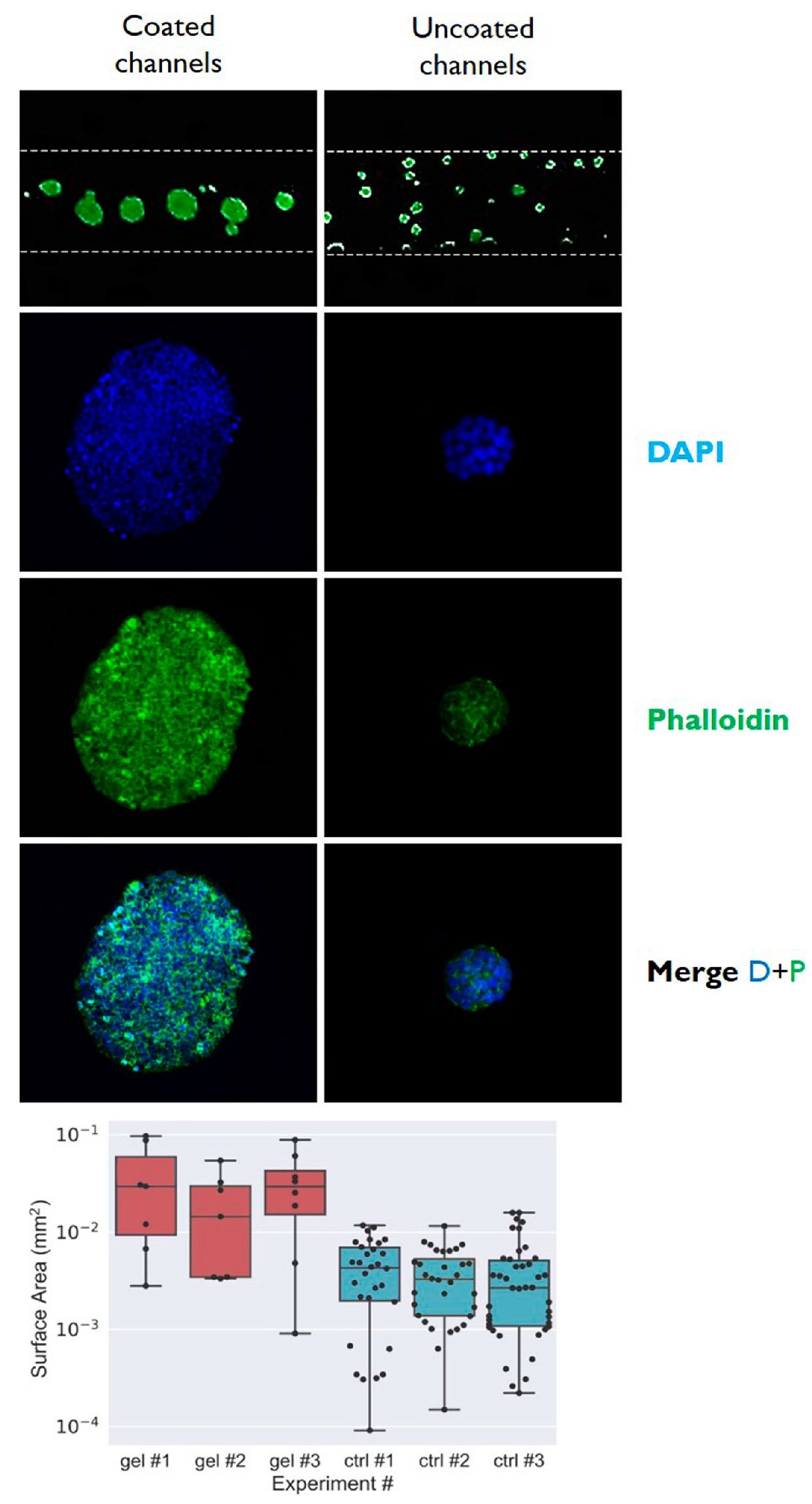
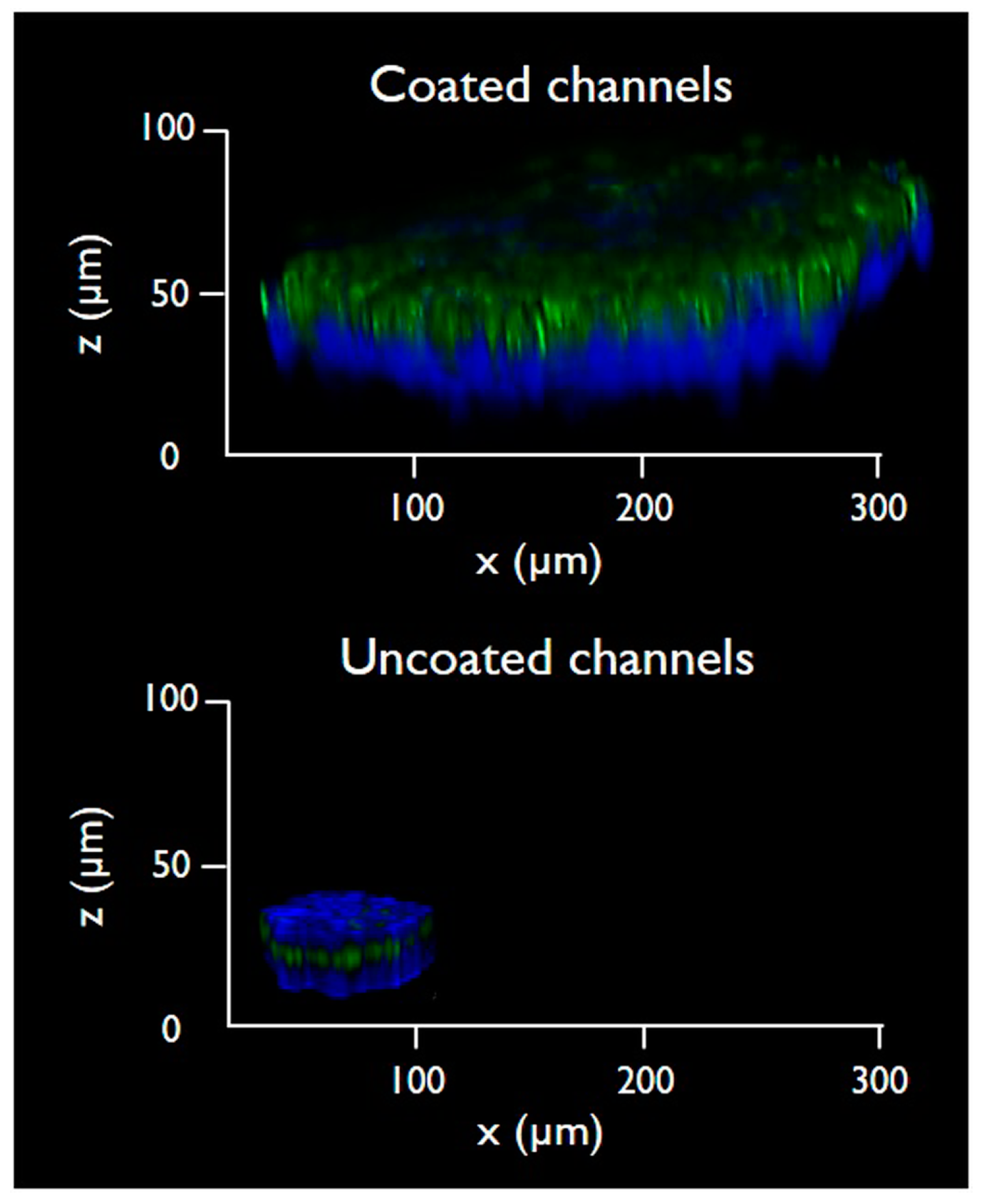
3.1. Fabrication of Gelatin-Coated Microfluidic Channels
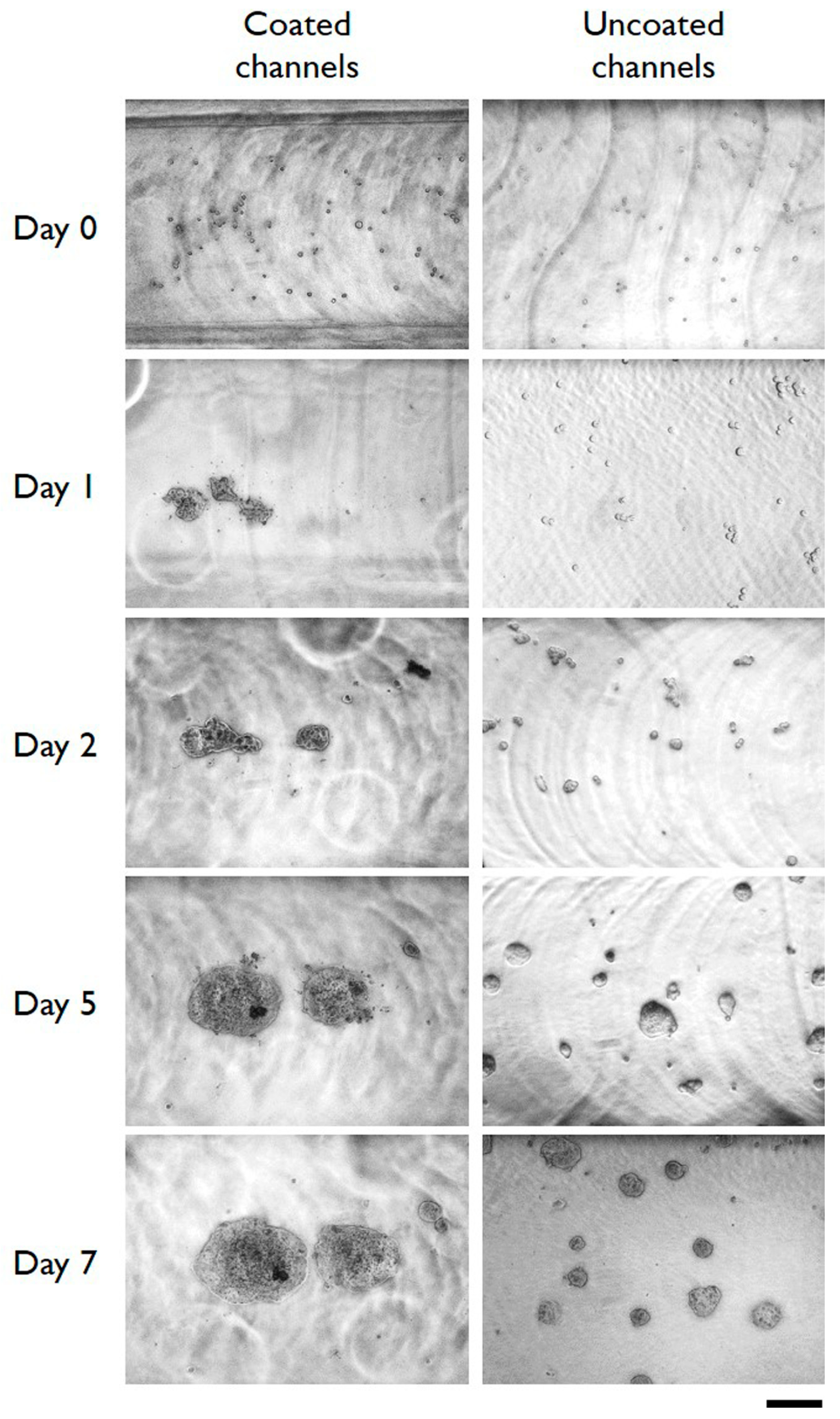
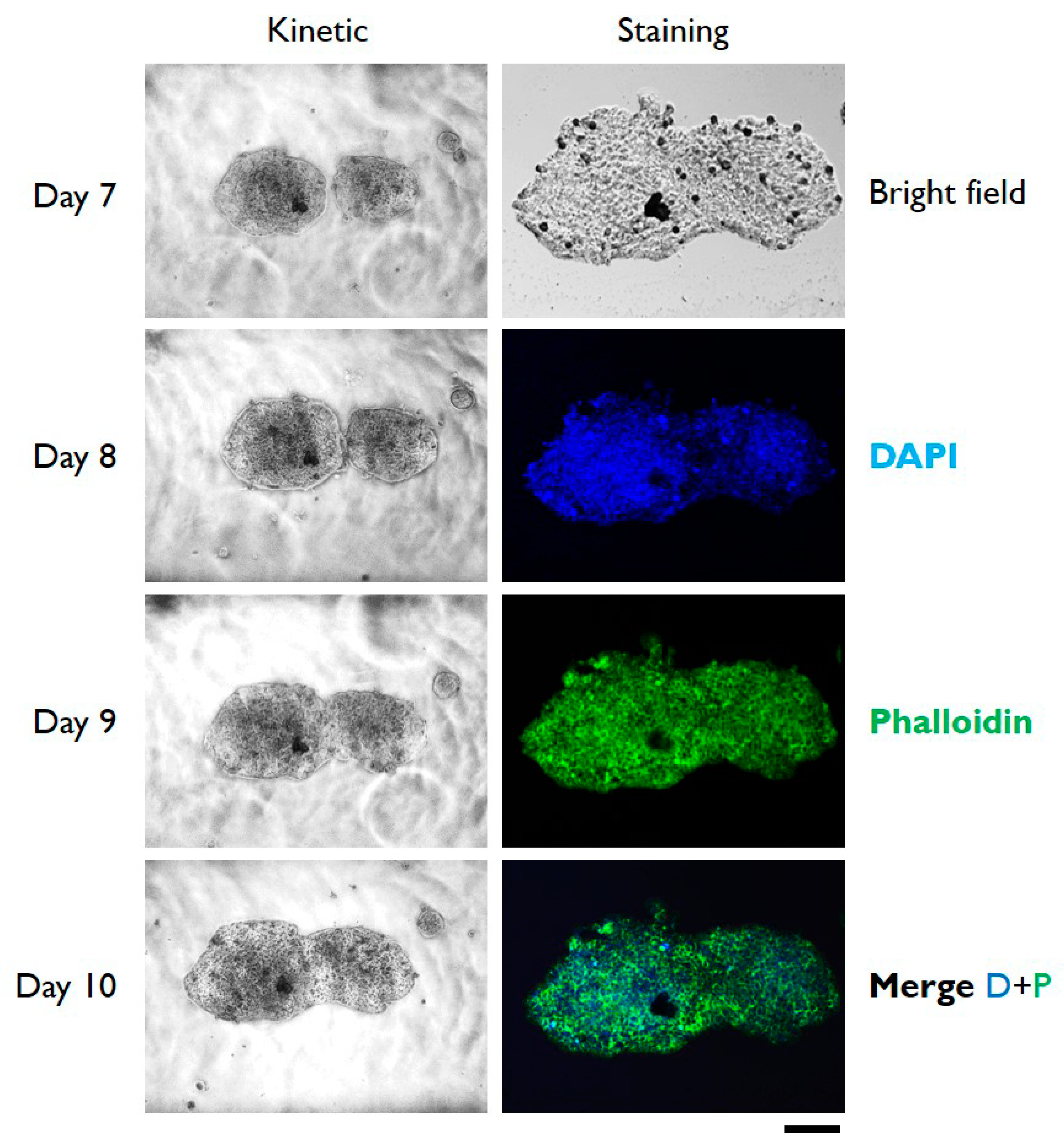
3.2. Cells Aggregation and 3D Microtissue Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef]
- Dolznig, H.; Rupp, C.; Puri, C.; Haslinger, C.; Schweifer, N.; Wieser, E.; Kerjaschki, D.; Garin-Chesa, P. Modeling colon adenocarcinomas in vitro: A 3D co-culture system induces cancer-relevant pathways upon tumor cell and stromal fibroblast interaction. Am. J. Pathol. 2011, 179, 487–501. [Google Scholar] [CrossRef]
- Fischbach, C.; Kong, H.J.; Hsiong, S.X.; Evangelista, M.B.; Yuen, W.; Mooney, D.J. Cancer cell angiogenic capability is regulated by 3D culture and integrin engagement. Proc. Natl. Acad. Sci. USA 2009, 106, 399–404. [Google Scholar] [CrossRef] [Green Version]
- Pickl, M.; Ries, C. Comparison of 3D and 2D tumor models reveals enhanced HER2 activation in 3D associated with an increased response to trastuzumab. Oncogene 2009, 28, 461. [Google Scholar] [CrossRef] [PubMed]
- Takagi, A.; Watanabe, M.; Ishii, Y.; Morita, J.; Hirokawa, Y.; Matsuzaki, T.; Shiraishi, T. Three-dimensional cellular spheroid formation provides human prostate tumor cells with tissue-like features. Anticancer Res. 2007, 27, 45–53. [Google Scholar]
- Desoize, B.; Jardillier, J.-C. Multicellular resistance: A paradigm for clinical resistance? Crit. Rev. Oncol./Hematol. 2000, 36, 193–207. [Google Scholar] [CrossRef]
- Brancato, V.; Gioiella, F.; Profeta, M.; Imparato, G.; Guarnieri, D.; Urciuolo, F.; Melone, P.; Netti, P.A. 3D tumor microtissues as an in vitro testing platform for microenvironmentally-triggered drug delivery systems. Acta Biomater. 2017, 57, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas 2. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar]
- Gong, X.; Lin, C.; Cheng, J.; Su, J.; Zhao, H.; Liu, T.; Wen, X.; Zhao, P. Generation of multicellular tumor spheroids with microwell-based agarose scaffolds for drug testing. PLoS ONE 2015, 10, e0130348. [Google Scholar] [CrossRef]
- Santo, V.E.; Estrada, M.F.; Rebelo, S.P.; Abreu, S.; Silva, I.; Pinto, C.; Veloso, S.C.; Serra, A.T.; Boghaert, E.; Alves, P.M.; et al. Adaptable stirred-tank culture strategies for large scale production of multicellular spheroid-based tumor cell models. J. Biotechnol. 2016, 221, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Timmins, N.E.; Nielsen, L.K. Generation of multicellular tumor spheroids by the hanging-drop method. Tissue Eng. 2007, 140, 141–151. [Google Scholar]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef]
- Vadivelu, R.K.; Kamble, H.; Shiddiky, M.J.; Nguyen, N.-T. Microfluidic Technology for the Generation of Cell Spheroids and Their Applications. Micromachines 2017, 8, 94. [Google Scholar] [CrossRef]
- Pitingolo, G.; Nizard, P.; Riaud, A.; Taly, V. Beyond the on/off Chip Trade-off: A Reversibly Sealed Microfluidic Platform for 3D Tumor Microtissue Analysis. Sens. Actuators B 2018, 274, 393–401. [Google Scholar] [CrossRef]
- Verhulsel, M.; Vignes, M.; Descroix, S.; Malaquin, L.; Vignjevic, D.M.; Viovy, J.-L. A review of microfabrication and hydrogel engineering for micro-organs on chips. Biomaterials 2014, 35, 1816–1832. [Google Scholar] [CrossRef] [PubMed]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef]
- Loebel, C.; Szczesny, S.E.; Cosgrove, B.D.; Alini, M.; Zenobi-Wong, M.; Mauck, R.L.; Eglin, D. Cross-linking chemistry of tyramine-modified hyaluronan hydrogels alters mesenchymal stem cell early attachment and behavior. Biomacromolecules 2017, 18, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Khademhosseini, A.; Langer, R. Microengineered hydrogels for tissue engineering. Biomaterials 2007, 28, 5087–5092. [Google Scholar] [CrossRef]
- Wang, L.; Lu, G.; Lu, Q.; Kaplan, D.L. Controlling Cell Behavior on Silk Nanofiber Hydrogels with Tunable Anisotropic Structures. ACS Biomater. Sci. Eng. 2018, 4, 933–941. [Google Scholar] [CrossRef]
- Chung, B.G.; Lee, K.-H.; Khademhosseini, A.; Lee, S.-H. Microfluidic fabrication of microengineered hydrogels and their application in tissue engineering. Lab Chip 2012, 12, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Husmann, A.; Hume, R.D.; Watson, C.J.; Cameron, R.E. Development of three-dimensional collagen scaffolds with controlled architecture for cell migration studies using breast cancer cell lines. Biomaterials 2017, 114, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.; Truong, N.F.; Segura, T. Design of cell–matrix interactions in hyaluronic acid hydrogel scaffolds. Acta Biomater. 2014, 10, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement membrane matrix with biological activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Camci-Unal, G.; Cuttica, D.; Annabi, N.; Demarchi, D.; Khademhosseini, A. Synthesis and characterization of hybrid hyaluronic acid-gelatin hydrogels. Biomacromolecules 2013, 14, 1085–1092. [Google Scholar] [CrossRef]
- Rose, J.; Pacelli, S.; Haj, A.; Dua, H.; Hopkinson, A.; White, L.; Rose, F. Gelatin-based materials in ocular tissue engineering. Materials 2014, 7, 3106–3135. [Google Scholar] [CrossRef] [PubMed]
- Speer, D.P.; Chvapil, M.; Eskelson, C.; Ulreich, J. Biological effects of residual glutaraldehyde in glutaraldehyde-tanned collagen biomaterials. J. Biomed. Mater. Res. 1980, 14, 753–764. [Google Scholar] [CrossRef]
- Benton, J.A.; DeForest, C.A.; Vivekanandan, V.; Anseth, K.S. Photocrosslinking of gelatin macromers to synthesize porous hydrogels that promote valvular interstitial cell function. Tissue Eng. Part A 2009, 15, 3221–3230. [Google Scholar] [CrossRef]
- Bae, H.; Ahari, A.F.; Shin, H.; Nichol, J.W.; Hutson, C.B.; Masaeli, M.; Kim, S.H.; Aubin, H.; Yamanlar, S.; Khademhosseini, A. Cell-laden microengineered pullulan methacrylate hydrogels promote cell proliferation and 3D cluster formation. Soft Matter 2011, 7, 1903–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Y.; Liu, X.; Wei, D.; Sun, J.; Xiao, W.; Zhao, H.; Guo, L.; Wei, Q.; Fan, H.; Zhang, X. Photo-cross-linkable methacrylated gelatin and hydroxyapatite hybrid hydrogel for modularly engineering biomimetic osteon. ACS Appl. Mater. Interfaces 2015, 7, 10386–10394. [Google Scholar] [CrossRef]
- Zhao, X.; Lang, Q.; Yildirimer, L.; Lin, Z.Y.; Cui, W.; Annabi, N.; Ng, K.W.; Dokmeci, M.R.; Ghaemmaghami, A.M.; Khademhosseini, A. Photocrosslinkable gelatin hydrogel for epidermal tissue engineering. Adv. Healthc. Mater. 2016, 5, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Yannas, A.; Tobolsky, A. Cross-Linking of gelatine by dehydration. Nature 1967, 215, 509. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, R.; Pitingolo, G.; Falanga, A.P.; Guarnieri, D.; Netti, P.A. Confined gelatin dehydration as a viable route to go beyond micromilling resolution and miniaturize biological assays. ACS Appl. Mater. Interfaces 2016, 8, 12075–12081. [Google Scholar] [CrossRef]
- Pitingolo, G.; Taly, V.; Nastruzzi, C. A Second Life for Old Electronic Parts: A Spin Coater for Microfluidic Applications; Royal Soc Chemistry: Cambridge, UK, 2018. [Google Scholar]
- Tsao, C.-W.; DeVoe, D.L. Bonding of thermoplastic polymer microfluidics. Microfluid. Nanofluid. 2009, 6, 1–16. [Google Scholar] [CrossRef]
- Brouzes, E.; Medkova, M.; Savenelli, N.; Marran, D.; Twardowski, M.; Hutchison, J.B.; Rothberg, J.M.; Link, D.R.; Perrimon, N.; Samuels, M.L. Droplet microfluidic technology for single-cell high-throughput screening. Proc. Natl. Acad. Sci. USA 2009, 106, 14195–14200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidenko, N.; Schuster, C.F.; Bax, D.V.; Farndale, R.W.; Hamaia, S.; Best, S.M.; Cameron, R.E. Evaluation of cell binding to collagen and gelatin: A study of the effect of 2D and 3D architecture and surface chemistry. J. Mater. Sci. Mater. Med. 2016, 27, 148. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Day | Semi-Major Axis Coated (µm) | Semi-Minor Axis Coated (µm) | Semi-Major Axis Uncoated (µm) | Semi-Minor Axis Uncoated (µm) | Total Area Coated (mm2) | Total Area Uncoated (mm2) |
|---|---|---|---|---|---|---|
| 7 | 201 ± 20 | 153 ± 12 | 50 ± 10 | 41 ± 7 | 0.965 | 0.064 |
| 8 | 216 ± 24 | 162 ± 18 | 53 ± 8 | 43 ± 4 | 1.098 | 0.071 |
| 9 | 249 ± 18 | 165 ± 26 | 60 ± 12 | 47 ± 9 | 1.290 | 0.088 |
| 10 | 262 ± 24 | 172 ± 9 | 72 ± 9 | 54 ± 11 | 1.415 | 0.122 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitingolo, G.; Riaud, A.; Nastruzzi, C.; Taly, V. Gelatin-Coated Microfluidic Channels for 3D Microtissue Formation: On-Chip Production and Characterization. Micromachines 2019, 10, 265. https://doi.org/10.3390/mi10040265
Pitingolo G, Riaud A, Nastruzzi C, Taly V. Gelatin-Coated Microfluidic Channels for 3D Microtissue Formation: On-Chip Production and Characterization. Micromachines. 2019; 10(4):265. https://doi.org/10.3390/mi10040265
Chicago/Turabian StylePitingolo, Gabriele, Antoine Riaud, Claudio Nastruzzi, and Valerie Taly. 2019. "Gelatin-Coated Microfluidic Channels for 3D Microtissue Formation: On-Chip Production and Characterization" Micromachines 10, no. 4: 265. https://doi.org/10.3390/mi10040265