Whole Blood Based Multiparameter Assessment of Thrombus Formation in Standard Microfluidic Devices to Proxy In Vivo Haemostasis and Thrombosis

 and
and
Abstract
:1. Introduction
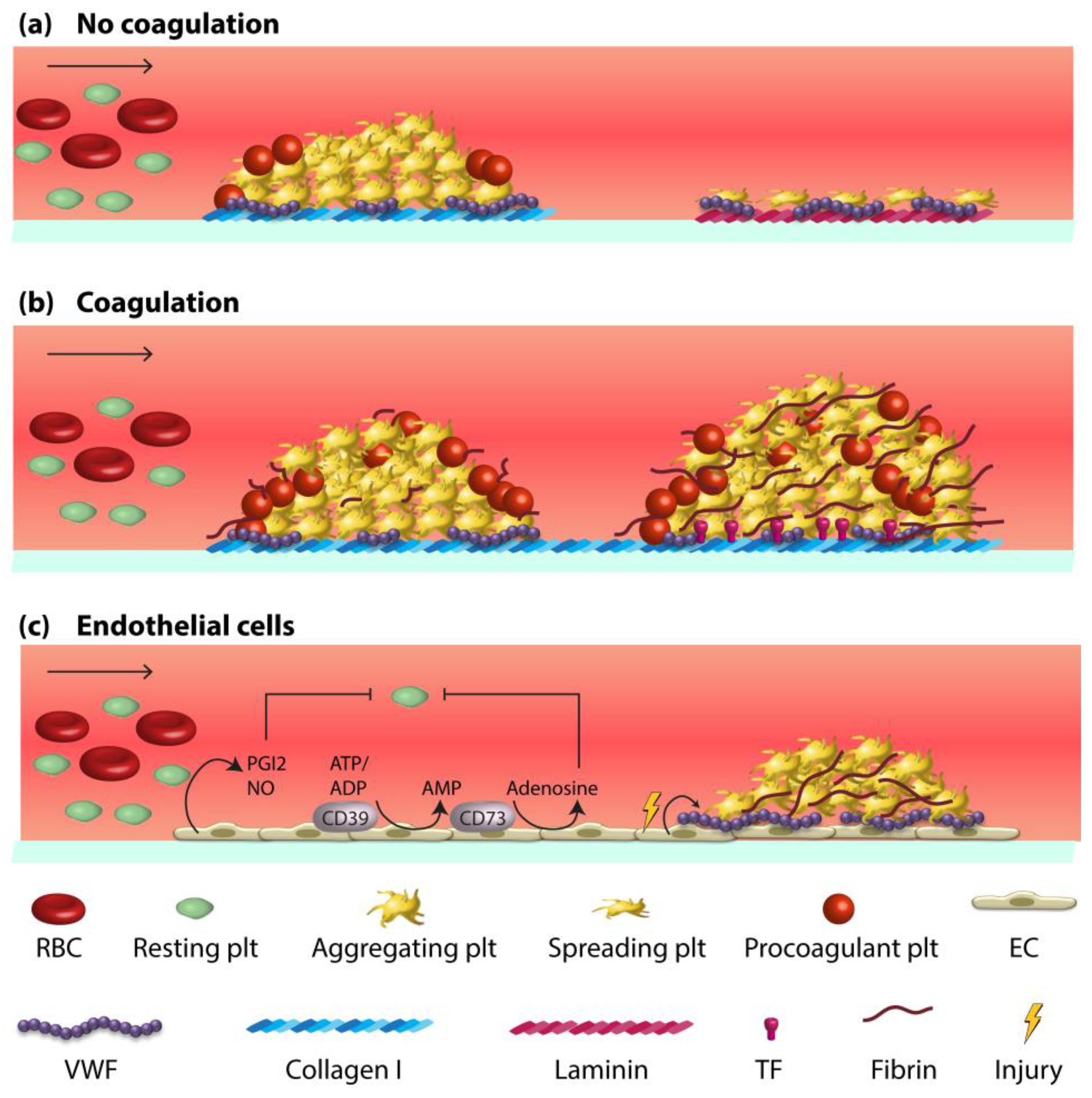
2. Vessel Wall-Blood Component Interactions in Haemostasis and Thrombosis
3. Whole Blood Microfluidics to Investigate and Measure Platelet Activation
4. Whole Blood Microfluidics to Study Platelet and Coagulation Activation
5. Whole Blood Microfluidics to Study Fibrinolysis
6. Microfluidics to Study Whole Blood Interactions with Endothelial Cells
7. Towards Standardisation and Clinical Use
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A
Appendix B

{kind=link}
{kind=link}
{kind=link}
| Device | Geometry | Shear rate | Anticoagulant | Sample | Thrombogenic Surface | Protocol | Coag. | Ref. |
|---|---|---|---|---|---|---|---|---|
| Maastricht flow chamber | d = 50 μm, w = 3 mm, l = 30 mm | 150 s−1, 1000 s−1, 1600 s−1 | Citrate, PPACK, fragmin | Whole blood | 52 surfaces | 4–6 min perfusion, image acquisition, staining, acquisition | No | [30] |
| Maastricht flow chamber | d = 50 μm, w = 3 mm, l = 30 mm | 1000 s−1 | Citrate | Whole blood | 6 surfaces | 3.5 min perfusion, staining, image acquisition, rinsing, acquisition | No | [32] |
| Parallel channels with stenoses | d = 0.18 mm, w = 0.2–1 mm, l = 70 mm | Calculated | Citrate, PPACK | Whole blood | VWF, fibrinogen | 5 min perfusion, rinsing, fixation, image acquisition | No | [78] |
| PDMS, 8 channels flow device | n.i. | 100 s−1 | CTI | Whole blood | Collagen, TF | 15 min perfusion, image acquisition every min | Yes | [79] |
| PDMS, 8 channels flow device | d = 60 μm, w = 250 μm | 200 s−1 | PPACK | Whole blood | Collagen | 5 min perfusion, image acquisition | No | [80] |
| Parallel plate flow chamber | d = 120 µm, w = 450 µm, l = 2 cm | 300 s−1 | Heparin | Whole blood | Collagen | 3 min perfusion, rinsing, image acquisition | No | [81] |
| PDMS ladder network | Main channels: w = 100 µm, d = 100 µm, Bypass channels: w = 50 | Variable | Citrate | Whole blood | Collagen | 30 min perfusion, rinsing, image acquisition | No | [82] |
| Maastricht flow chamber | d = 50 μm, w = 3 mm, l = 30 mm | 150 s−1, 500 s−1, 1000 s−1 | Citrate | Whole blood | Collagen/TF, plaque material | 10 min recalcified, image acquisition every 2 min | Yes | [13,83,84] |
| PDMS vs. Ibidi sticky-slide I 0.1 | PDMS: d = 60 μm, w = 250 μm vs. Ibidi | 1400 s−1 | Hirudin | n.i. | Collagen | Perfusion, image acquisition | No | [85] |
| Ibidi μ-slide-I 0.1 Luer | d = 100 μm, w = 5 mm, l = 60 mm | 1500 s−1 | n.i. | PRP | Collagen | 5 min perfusion, rinsing, image acquisition | No | [86] |
| PDMS, 8 channels flow device | d = 60 μm, w = 250 μm | 100 s−1, 1000 s−1 | CTI | Whole blood | Collagen ± TF, VWF | Perfusion, image acquisition | Yes | [87] |
| Laser cut PSA | d = 50 μm, w = 2 mm, l = 75 mm | 1500 s−1 | Citrate | Whole blood | VWF | Perfusion, image acquisition | No | [88] |
| Well plate device | n.i. | 250 s−1, 5000 s−1 | Citrate | Whole blood | Collagen, VWF | 2–5 min perfusion, image acquisition | No | [89] |
References
- Baumgartner, H.R.; Muggli, R.; Tschopp, T.B.; Turitto, V.T. Platelet adhesion, release and aggregation in flowing blood: Effects of surface properties and platelet function. Thromb. Haemost. 1976, 35, 124–138. [Google Scholar] [CrossRef]
- Bolhuis, P.A.; Sakariassen, K.S.; Sixma, J.J. Adhesion of blood platelets to human arterial subendothelium: Role of factor VIII-Von Willebrand factor. Haemostasis 1979, 8, 312–323. [Google Scholar] [CrossRef]
- Badimon, L.; Badimon, J.J.; Galvez, A.; Chesebro, J.H.; Fuster, V. Influence of arterial damage and wall shear rate on platelet deposition. Ex vivo study in a swine model. Atherosclerosis 1986, 6, 312–320. [Google Scholar] [CrossRef]
- Brouns, S.L.; van Geffen, J.P.; Heemskerk, J.W. High-Throughput measurement of human platelet aggregation under flow: Application in hemostasis and beyond. Platelets 2018, 29, 662–669. [Google Scholar] [CrossRef]
- Sakariassen, K.S.; Hanson, S.R.; Cadroy, Y. Methods and models to evaluate shear-dependent and surface reactivity-dependent antithrombotic efficacy. Thromb. Res. 2001, 104, 149–174. [Google Scholar] [CrossRef]
- Baaten, C.C.; Meacham, S.; de Witt, S.M.; Feijge, M.A.; Adams, D.J.; Akkerman, J.W.; Cosemans, J.M.; Grassi, L.; Jupe, S.; Kostadima, M.; et al. A synthesis approach of mouse studies to identify genes and proteins in arterial thrombosis and bleeding. Blood 2018, 132, e35–e46. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef]
- Van der Meijden, P.E.; Heemskerk, J.W. Platelet biology and functions: New concepts and future clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar]
- Van der Meijden, P.E.; Munnix, I.C.; Auger, J.M.; Govers-Riemslag, J.W.; Cosemans, J.M.; Kuijpers, M.J.; Spronk, H.M.; Watson, S.P.; Renné, T.; Heemskerk, J.W. Dual role of collagen in factor XII-dependent thrombus and clot formation. Blood 2009, 114, 881–890. [Google Scholar] [CrossRef]
- Heemskerk, J.W.; Bevers, E.M.; Lindhout, T. Platelet activation and blood coagulation. Thromb. Haemost. 2002, 88, 186–193. [Google Scholar]
- Siljander, P.M.; Farndale, R.W.; Feijge, M.A.; Comfurius, P.; Kos, S.; Bevers, E.M.; Heemskerk, J.W. Platelet adhesion enhances the glycoprotein VI–dependent procoagulant response: Involvement of p38 MAP kinase and calpain. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 618–627. [Google Scholar] [CrossRef]
- De Witt, S.; Verdoold, R.; Cosemans, J.M.; Heemskerk, J.W. Insights into platelet-based control of coagulation. Thromb. Res. 2014, 133, S139–S148. [Google Scholar] [CrossRef]
- Swieringa, F.; Baaten, C.C.; Mastenbroek, T.; Rijnveld, N.; van der Laan, K.; Collins, P.; Lancé, M.D.; Henskens, Y.; Cosemans, J.M.; Verdoold, R.; et al. Platelet control of fibrin distribution and microelasticity in thrombus formation under flow. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 692–699. [Google Scholar] [CrossRef]
- Weiss, H.J.; Turitto, V.T. Prostacyclin (prostaglandin I2, PGI2) inhibits platelet adhesion and thrombus formation on subendothelium. Blood 1979, 53, 244–250. [Google Scholar] [CrossRef]
- Van IJzendoorn, S.C.; Heemskerk, J.W.; Reutelingsperger, C.P. Interactions between endothelial cells and blood Platelets. Endothelium 1995, 3, 81–98. [Google Scholar] [CrossRef]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 1987, 330, 1057–1058. [Google Scholar] [CrossRef]
- Marcus, A.J.; Broekman, M.J.; Drosopoulos, J.H.; Pinsky, D.J.; Islam, N.; Maliszewsk, C.R. Inhibition of platelet recruitment by endothelial cell CD39/ecto-ADPase: Significance for occlusive vascular diseases. Ital. Heart J. 2001, 2, 824–830. [Google Scholar]
- Wu, C.; Kim, P.Y.; Swystun, L.L.; Liaw, P.C.; Weitz, J.I. Activation of protein C and thrombin activable fibrinolysis inhibitor on cultured human endothelial cells. J. Thromb. Haemost. 2016, 14, 366–374. [Google Scholar] [CrossRef]
- Coenen, D.M.; Mastenbroek, T.G.; Cosemans, J.M. Platelet interaction with activated endothelium: Mechanistic insights from microfluidics. Blood 2017, 130, 2819–2828. [Google Scholar] [CrossRef]
- Kroll, M.H.; Hellums, J.D.; McIntire, L.V.; Schafer, A.I.; Moake, J.L. Platelets and shear stress. Blood 1996, 88, 1525–1541. [Google Scholar] [CrossRef]
- Nagy, M.; Heemskerk, J.W.; Swieringa, F. Use of microfluidics to assess the platelet-based control of coagulation. Platelets 2017, 28, 441–448. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, J.; Lopez, J.A. Flow-Driven assembly of VWF fibres and webs in in vitro microvessels. Nat. Commun. 2015, 6, 7858. [Google Scholar] [CrossRef]
- Ruggeri, Z.M.; Dent, J.A.; Saldívar, E. Contribution of distinct adhesive interactions to platelet aggregation in flowing blood. Blood 1999, 94, 172–178. [Google Scholar] [CrossRef]
- Ruggeri, Z.M. Mechanisms initiating platelet thrombus formation. Thromb. Haemost. 1997, 78, 611–616. [Google Scholar] [CrossRef]
- Konstantopoulos, K.; Grotta, J.C.; Sills, C.; Wu, K.K.; Hellums, J.D. Shear-Induced platelet aggregation in normal subjects and stroke patients. Thromb. Haemost. 1995, 74, 1329–1334. [Google Scholar] [CrossRef]
- Goto, S.; Sakai, H.; Goto, M.; Ono, M.; Ikeda, Y.; Handa, S.; Ruggeri, Z.M. Enhanced shear-induced platelet aggregation in acute myocardial infarction. Circulation 1999, 99, 608–613. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; Topper, J.N.; Nagel, T.; Anderson, K.R.; Garcia-Cardeña, G. Endothelial dysfunction, hemodynamic forces, and atherogenesis. Ann. N. Y. Acad. Sci. 2000, 902, 230–239. [Google Scholar] [CrossRef]
- Westein, E.; de Witt, S.; Lamers, M.M.; Cosemans, J.M.; Heemskerk, J.W. Monitoring in vitro thrombus formation with novel microfluidic devices. Platelets 2012, 23, 501–509. [Google Scholar] [CrossRef]
- Van Kruchten, R.; Cosemans, J.M.; Heemskerk, J.W. Measurement of whole blood thrombus formation using parallel-plate flow chambers: A practical guide. Platelets 2012, 23, 229–242. [Google Scholar] [CrossRef]
- De Witt, S.M.; Swieringa, F.; Cavill, R.; Lamers, M.M.; van Kruchten, R.; Mastenbroek, T.; Baaten, C.; Coort, S.; Pugh, N.; Schulz, A.; et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat. Commun. 2014, 5, 4257. [Google Scholar] [CrossRef]
- Nagy, M.; Mastenbroek, T.G.; Mattheij, N.J.; De Witt, S.; Clemetson, K.J.; Kirschner, J.; Schulz, A.; Braun, A.; Cosemans, J.M.; Zieger, B.; et al. Variable impairment of platelet functions in patients with severe, genetically linked immune deficiencies. Haematologica 2018, 103, 540–549. [Google Scholar] [CrossRef]
- Van Geffen, J.P.; Brouns, S.; Batista, J.; McKinney, H.; Kempster, C.; Sivapalaratnam, S.; Baaten, C.B.; Boury, N.; Frontini, M.; Nagy, M.; et al. High-Throughput elucidation of thrombus formation reveals sources of platelet function variability. Haematologica 2019, 104, 1256–1267. [Google Scholar] [CrossRef]
- Petersen, R.; Lambourne, J.J.; Javierre, B.M.; Grassi, L.; Kreuzhuber, R.; Ruklisa, D.; Rosa, I.M.; Tome, R.A.; Elding, H.; van Geffen, J.P.; et al. Platelet function is modified by common sequence variation in megakaryocyte super enhancer. Nat. Commun. 2017, 8, 16058. [Google Scholar] [CrossRef]
- Von Hundelshausen, P.; Agten, S.M.; Eckardt, V.; Blanchet, X.; Schmitt, M.M.; Ippel, H.; Neideck, C.; Bidzhekov, K.; Leberzammer, J.; Wichapong, K.; et al. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl. Med. 2017, 9, 384. [Google Scholar] [CrossRef]
- Münzer, P.; Borst, O.; Walker, B.; Schmid, E.; Feijge, M.A.; Cosemans, J.M.; Chatterjee, M.; Schmidt, E.M.; Schmidt, S.; Towhid, S.T.; et al. Acid sphingomyelinase regulates platelet cell membrane scrambling, secretion, and thrombus formation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, N.; Seijkens, T.; Lievens, D.; Kuijpers, M.J.; Winkels, H.; Projahn, D.; Hartwig, H.; Beckers, L.; Megens, R.T.; Boon, L.; et al. Platelet CD40 exacerbates atherosclerosis by transcellular activation of endothelial cells and leukocytes. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Lepropre, S.; Kautbally, S.; Octave, M.; Ginion, A.; Onselaer, M.B.; Steinberg, G.R.; Kemp, B.E.; Hego, A.; Wera, O.; Brouns, S.; et al. AMPK-ACC signaling modulates platelet phospholipids content and potentiates platelet function and thrombus formation. Blood 2018, 132, 1180–1192. [Google Scholar] [CrossRef]
- Gotru, S.K.; van Geffen, J.P.; Nagy, M.; Mammadova-Bach, E.; Eilenberger, J.; Volz, J.; Manukjan, G.; Schulze, H.; Eber, S.; Schambeck, C.; et al. Defective Zn2+ homeostasis in mouse and human platelets with α- and δ-storage pool diseases. Sci. Rep. 2019, 9, 8333. [Google Scholar] [CrossRef]
- Nagy, M.; van Geffen, J.P.; Stegner, D.; Adams, D.; Braun, A.; de Witt, S.M.; Elvers, M.; Kuijpers, M.J.; Kunzelmann, K.; Oury, C.; et al. Comparative analysis of microfluidics thrombus formation in multiple genetically modified mice: Link to thrombosis and hemostasis. Front. Cardiovasc. Med. 2019, 6, 99. [Google Scholar] [CrossRef] [Green Version]
- Zwaginga, J.J.; Nash, G.; King, M.R.; Heemskerk, J.W.; Frojmovic, M.; Hoylaerts, M.; Sakariassen, K.S. Flow-Based assays for global assessment of haemostasis. Part 1: Biorheologic considerations. J. Thromb. Haemost. 2006, 4, 2486–2487. [Google Scholar] [CrossRef]
- Zwaginga, J.J.; Sakariassen, K.S.; Nash, G.; King, M.; Heemskerk, J.W.; Frojmovic, M.; Hoylaerts, M.F. Flow-Based assays for global assessment of haemostasis. Part 2: Current methods and considerations for the future. J. Thromb. Haemost. 2006, 4, 2716–2717. [Google Scholar] [CrossRef]
- Roest, M.; Reininger, A.; Zwaginga, J.J.; King, M.R.; Heemskerk, J.W. Flow chamber-based assays to measure thrombus formation in vitro: Requirements for standardization. J. Thromb. Haemost. 2011, 9, 2322–23224. [Google Scholar] [CrossRef]
- Van Geffen, J.P.; Swieringa, F.; Heemskerk, J.W. Platelets and coagulation in thrombus formation: Aberrations in the Scott syndrome. Thromb. Res. 2016, 141, S12–S16. [Google Scholar] [CrossRef]
- Colace, T.V.; Muthard, R.W.; Diamond, S.L. Thrombus growth and embolism on tissue factor-bearing collagen surfaces under flow: Role of thrombin with and without fibrin. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Pugh, N.; Jarvis, G.; Koch, A.; Sakariassen, K.S.; Davis, B.; Farndale, R.W. The impact of factor Xa inhibition on axial dependent arterial thrombus formation triggered by a tissue factor rich surface. J. Thromb. Thrombolysis. 2012, 33, 6–15. [Google Scholar] [CrossRef]
- Colace, T.V.; Tormoen, G.W.; McCarty, O.J.; Diamond, S.L. Microfluidics and coagulation biology. Annu. Rev. Biomed. Eng. 2013, 15, 283–303. [Google Scholar] [CrossRef] [Green Version]
- Schoeman, R.M.; Rana, K.; Danes, N.; Lehmann, M.; Di Paola, J.A.; Fogelson, A.L.; Leiderman, K.; Neeves, K.B. A microfluidic model of hemostasis sensitive to platelet function and coagulation. Cell. Mol. Bioengin. 2017, 10, 3–15. [Google Scholar] [CrossRef]
- Westein, E.; van der Meer, A.D.; Kuijpers, M.J.; Frimat, J.P.; van den Berg, A.; Heemskerk, J.W. Atherosclerotic geometries spatially confine and exacerbate pathological thrombus formation poststenosis in a von Willebrand factor-dependent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 1357–1362. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.V.; Standeven, K.F.; Ariëns, R.A. Fibrinogen gamma-chain splice variant gamma’ alters fibrin formation and structure. Blood 2003, 102, 535–540. [Google Scholar] [CrossRef]
- Wolberg, A.S. Thrombin generation and fibrin clot structure. Blood Rev. 2007, 21, 131–142. [Google Scholar] [CrossRef]
- Whyte, C.S.; Swieringa, F.; Mastenbroek, T.G.; Lionikiene, A.S.; Lancé, M.D.; van der Meijden, P.E.; Heemskerk, J.W.; Mutch, N.J. Plasminogen associates with phosphatidylserine-exposing platelets and contributes to thrombus lysis under flow. Blood 2015, 125, 2568–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schols, S.E.; Lancé, M.D.; Feijge, M.A.; Damoiseaux, J.; Marcus, M.; Hamulyák, K.; ten Cate, H.; Heemskerk, J.W.; van Pampus, E.C. Impaired thrombin generation and fibrin clot formation in patients with dilutional coagulopathy during major surgery. Thromb. Haemost. 2010, 103, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Swieringa, F.; Kuijpers, M.J.; Lamers, M.M.; van der Meijden, P.E.; Heemskerk, J.W. Rate-Limiting roles of tenase complex of factors VIII and IX in platelet procoagulant activity and formation of platelet-fibrin thrombi under flow. Haematologica 2016, 100, 748–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, P.F.; Albers, H.J.; Linssen, J.E.; Middelkamp, H.H.; van der Hout, L.; Passier, R.; van den Berg, A.; Malda, J.; van der Meer, A.D. Mimicking arterial thrombosis in a 3D-printed microfluidic in vitro vascular model based on computed tomography angiography data. Lab. Chip. 2017, 17, 2785–2792. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, M.S.; Palange, A.L.; Mancuso, A.; Grande, L.; Muccari, D.; Scavelli, F.B.; Irace, C.; Gnasso, A.; Carallo, C. Decreased platelet aggregation by shear stress-stimulated endothelial cells in vitro: Description of a method and first results in diabetes. Diabetes Vasc. Dis. Res. 2015, 12, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Garland, K.S.; Reitsma, S.E.; Shirai, T.; Zilberman-Rudenko, J.; Tucker, E.I.; Gailani, D.; Gruber, A.; McCarty, O.J.; Puy, C. Removal of the C-terminal domains of ADAMTS13 by activated coagulation factor XI induces platelet adhesion on endothelial cells under flow conditions. Front. Med. 2017, 4, 232. [Google Scholar] [CrossRef] [Green Version]
- Gogia, S.; Kelkar, A.; Zhang, C.; Dayananda, K.M.; Neelamegham, S. Role of calcium in regulating the intra- and extracellular cleavage of von Willebrand factor by the protease ADAMTS13. Blood Adv. 2017, 1, 2063–2074. [Google Scholar] [CrossRef] [Green Version]
- Meyer Dos Santos, S.; Blankenbach, K.; Scholich, K.; Dorr, A.; Monsefi, N.; Keese, M.; Linke, B.; Deckmyn, H.; Nelson, K.; Harder, S. Platelets from flowing blood attach to the inflammatory chemokine CXCL16 expressed in the endothelium of the human vessel wall. Thromb. Haemost. 2015, 114, 297–312. [Google Scholar] [CrossRef]
- Navarro-Nunez, L.; Pollitt, A.Y.; Lowe, K.; Latif, A.; Nash, G.B.; Watson, S.P. Platelet adhesion to podoplanin under flow is mediated by the receptor CLEC-2 and stabilised by Src/Syk-dependent platelet signalling. Thromb. Haemost. 2015, 113, 1109–1120. [Google Scholar]
- Popa, M.; Tahir, S.; Elrod, J.; Kim, S.H.; Leuschner, F.; Kessler, T.; Bugert, P.; Pohl, U.; Wagner, A.H.; Hecker, M. Role of CD40 and ADAMTS13 in von Willebrand factor-mediated endothelial cell-platelet-monocyte interaction. Proc. Natl. Acad. Sci. USA 2018, 115, E5556–E5565. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, Y.; Hardy, E.T.; Ahn, B.; Tran, R.; Fay, M.E.; Ciciliano, J.C.; Mannino, R.G.; Myers, D.R.; Qiu, Y.; Carden, M.A.; et al. A microengineered vascularized bleeding model that integrates the principal components of hemostasis. Nat. Commun. 2018, 9, 509. [Google Scholar] [CrossRef] [PubMed]
- Sylman, J.L.; Artzer, D.T.; Rana, K.; Neeves, K.B. A vascular injury model using focal heat-induced activation of endothelial cells. Integr. Biol. 2015, 7, 801–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchhofer, D.; Tschopp, T.B.; Hadváry, P.; Baumgartner, H.R. Endothelial cells stimulated with tumor necrosis factor-alpha express varying amounts of tissue factor resulting in inhomogenous fibrin deposition in a native blood flow system. Effects of thrombin inhibitors. J. Clin. Investig. 1994, 93, 2073–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; van der Meer, A.D.; Papa, A.L.; Barrile, R.; Lai, A.; Schlechter, B.L.; Otieno, M.A.; Louden, C.S.; Hamilton, G.A.; Michelson, A.D.; et al. Assessment of whole blood thrombosis in a microfluidic device lined by fixed human endothelium. Biomed. Microdevices 2016, 18, 73. [Google Scholar] [CrossRef] [Green Version]
- Michels, A.; Swystun, L.L.; Mewburn, J.; Albanez, S.; Lillicrap, D. Investigating von Willebrand factor pathophysiology using a flow chamber model of von Willebrand factor-platelet string formation. J. Vis. Exp. 2017, 126, 1. [Google Scholar] [CrossRef]
- Goerge, T.; Kleinerüschkamp, F.; Barg, A.; Schnaeker, E.M.; Huck, V.; Schneider, M.F.; Steinhoff, M.; Schneider, S.W. Microfluidic reveals generation of platelet-strings on tumor-activated endothelium. Thromb. Haemost. 2007, 98, 283–286. [Google Scholar]
- Dong, J.F.; Moake, J.L.; Nolasco, L.; Bernardo, A.; Arceneaux, W.; Shrimpton, C.N.; Schade, A.J.; McIntire, L.V.; Fujikawa, K.; López, J.A. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002, 100, 4033–4039. [Google Scholar] [CrossRef] [Green Version]
- Noone, D.G.; Riedl, M.; Pluthero, F.G.; Bowman, M.L.; Liszewski, M.K.; Lu, L.; Quan, Y.; Balgobin, S.; Schneppenheim, R.; Schneppenheim, S.; et al. Von Willebrand factor regulates complement on endothelial cells. Kidney Int. 2016, 90, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Barrile, R.; van der Meer, A.D.; Mammoto, A.; Mammoto, T.; De Ceunynck, K.; Aisiku, O.; Otieno, M.A.; Louden, C.S.; Hamilton, G.A.; et al. Primary human lung alveolus-on-a-chip model of intravascular thrombosis for assessment of therapeutics. Clin. Pharmacol. Ther. 2018, 103, 332–340. [Google Scholar] [CrossRef]
- Neeves, K.B.; Onasoga, A.A.; Hansen, R.R.; Lilly, J.J.; Venckunaite, D.; Sumner, M.B.; Irish, A.T.; Brodsky, G.; Manco-Johnson, M.J.; Di Paola, J.A. Sources of variability in platelet accumulation on type 1 fibrillar collagen in microfluidic flow assays. PLoS ONE 2013, 7, e54680. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, M.; Ashworth, K.; Manco-Johnson, M.; Di Paola, J.; Neeves, K.B.; Ng, C.J. Evaluation of a microfluidic flow assay to screen for von Willebrand disease and low von Willebrand factor levels. J. Thromb. Haemost. 2017, 16, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoeman, R.M.; Lehmann, M.; Neeves, K.B. Flow chamber and microfluidic approaches for measuring thrombus formation in genetic bleeding disorders. Platelets 2017, 28, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.; Mackie, I.; Mumford, A.; Briggs, C.; Liesner, R.; Winter, M.; Machin, S. British Committee for Standards in Haematology. Guidelines for the laboratory investigations of heritable disorders of platelet function. Br. J. Haematol. 2011, 155, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Dawood, B.B.; Lowe, G.C.; Lordkipanidze, M.; Bern, D.; Daly, M.E.; Makris, M.; Mumford, A.; Wilde, J.T.; Watson, S.P. Evaluation of participants with suspected heritable platelet function disorders including recommendation and validation of a streamlined agonst panel. Blood 2012, 120, 5041–5049. [Google Scholar] [CrossRef] [Green Version]
- Goodall, A.H.; Appleby, J. Flow-Cytometric analysis of platelet-membrane glycoprotein expression and platelet activation. Methods Mol. Biol. 2004, 272, 225–253. [Google Scholar]
- Hayward, C.P.; Harrison, P.; Cattaneo, M.; Ortel, T.L.; Rao, A.K. SSC-ISTH Platelet function analyzer (PFA)-100 closure time in the evaluation of platelet disorders and platelet function. J. Thromb. Haemost. 2006, 4, 312–319. [Google Scholar] [CrossRef]
- De Witt, S.; Swieringa, F.; Cosemans, J.M.; Heemkerk, J.W. Thrombus formation on microspotted arrays of thrombogenic surfaces. Nat. Protocol Exch. 2014, 2014, 3309. [Google Scholar]
- Rahman, S.M.; Hlady, V. Downstream platelet adhesion and activation under highly elevated upstream shear forces. Acta Biomater. 2019, 91, 135–143. [Google Scholar] [CrossRef]
- Li, R.; Panckeri, K.A.; Fogarty, P.F.; Cuker, A.; Diamond, S.L. Recombinant factor VIIa addition to haemophilic blood perfused over collagen/tissue factor can sufficiently bypass the factor IXa/VIIIa defect to rescue fibrin generation. Haemophilia 2017, 23, 759–768. [Google Scholar] [CrossRef]
- Li, R.; Diamond, S.L. Detection of platelet sensitivity to inhibitors of COX-1, P2Y₁, and P2Y₁₂ using a whole blood microfluidic flow assay. Thromb. Res. 2014, 133, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Jahn, K.; Suchodolski, K.; Schäfer, A.; Sahlmann, B.; Küster, U.; Echtermeyer, F.; Calmer, S.; Theilmeier, G.; Johanning, K. Effect of clopidogrel on thrombus formation in an ex vivo parallel plate flow chamber model cannot be reversed by addition of platelet concentrates or vWF concentrate. Anesth. Analg. 2017, 124, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Zilberman-Rudenko, J.; Sylman, J.L.; Lakshmanan, H.H.; McCarty, O.J.; Maddala, J. Dynamics of blood flow and thrombus formation in a multi-bypass microfluidic ladder network. Cell. Mol. Bioengin. 2017, 10, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Nergiz-Unal, R.; Cosemans, J.M.; Feijge, M.A.; van der Meijden, P.E.; Storey, R.F.; van Giezen, J.J.; oude Egbrink, M.G.; Heemskerk, J.W.; Kuijpers, M.J. Stabilizing role of platelet P2Y12 receptors in shear-dependent thrombus formation on ruptured plaques. PLoS ONE 2010, 5, e10130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijpers, M.J.; De Witt, S.; Nergiz-Unal, R.; van Kruchten, R.; Korporaal, S.J.; Febbraio, M.; Twja, M.; Voshol, P.J.; Hoylaerts, M.F.; Cosemans, J.M.; et al. Supporting roles of platelet thrombospondin-1 and CD36 in thrombus formation on collagen. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1187–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claesson, K.; Lindahl, T.L.; Faxolv, L. Counting the platelets: A robust and sensitive quantification method for thrombus formation. Thromb. Haemost. 2016, 115, 1178–1190. [Google Scholar] [PubMed]
- Van de Vijver, E.; De Cuyper, I.M.; Gerrits, A.J.; Verhoeven, A.J.; Seeger, K.; Gutiérrez, L.; van den Berg, T.K.; Kuijpers, T.W. Defects in Glanzmann thrombasthenia and LAD-III (LAD-1/v) syndrome: The role of integrin β1 and β3 in platelet adhesion to collagen. Blood 2012, 119, 583–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Tomaiuoloc, M.; Diamond, S.L. Minimum wound size for clotting: Flowing blood coagulates on a single collagen fiber presenting tissue factor and von Willebrand factor. Integr. Biol. 2016, 8, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Tovar-Lopez, F.J.; Rosengarten, G.; Westein, E.; Khoshmanesh, K.; Jackson, S.P.; Mitchell, A.; Nesbitt, W.S. A microfluidics device to monitor platelet aggregation dynamics in response to strain rate micro-gradients in flowing Blood. Lab. Chip. 2010, 10, 291–302. [Google Scholar] [CrossRef]
- Conant, C.G.; Nevill, J.T.; Zhou, Z.; Dong, J.F.; Schwartz, M.A.; Ionescu-Zanetti, C. Using well-plate microfluidic devices to conduct shear-based thrombosis assays. J. Lab. Autom. 2011, 16, 148–152. [Google Scholar] [CrossRef]



| Endothelial Injury | Surface | Shear Rate | Output Measurement | Ref. |
|---|---|---|---|---|
| Histamine, sCD40L, bradykinin; activated platelets | gelatin | 2.5–10 dyne/cm2 | platelets on large VWF multimers | [60,65] |
| TNF-α | not specified | 2.5 dyne/cm2 | platelets on-VWF strings | [56] |
| Histamine | not specified | 1–2.5 dyne/cm2 | VWF strings | [57] |
| Phorbol myristate acetate | rat tail collagen | 10–30 dyne/cm2 | VWF strings, platelet adhesion | [61,65] |
| Stenosis | rat tail collagen | 1000 s−1 | platelet aggregation | [54] |
| Mechanical injury | collagen type I | 500 or 2500 s−1 | platelets adhered to VWF | [61] |
| Tumor supernatant | gelatin | venous | ULVWF multimer secretion, VWF-platelet strings length | [66] |
| Histamine | gelatin | 2.5–50 dyne/cm2 | platelets on VWF-strings | [67] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Provenzale, I.; Brouns, S.L.N.; van der Meijden, P.E.J.; Swieringa, F.; Heemskerk, J.W.M. Whole Blood Based Multiparameter Assessment of Thrombus Formation in Standard Microfluidic Devices to Proxy In Vivo Haemostasis and Thrombosis. Micromachines 2019, 10, 787. https://doi.org/10.3390/mi10110787
Provenzale I, Brouns SLN, van der Meijden PEJ, Swieringa F, Heemskerk JWM. Whole Blood Based Multiparameter Assessment of Thrombus Formation in Standard Microfluidic Devices to Proxy In Vivo Haemostasis and Thrombosis. Micromachines. 2019; 10(11):787. https://doi.org/10.3390/mi10110787
Chicago/Turabian StyleProvenzale, Isabella, Sanne L. N. Brouns, Paola E. J. van der Meijden, Frauke Swieringa, and Johan W. M. Heemskerk. 2019. "Whole Blood Based Multiparameter Assessment of Thrombus Formation in Standard Microfluidic Devices to Proxy In Vivo Haemostasis and Thrombosis" Micromachines 10, no. 11: 787. https://doi.org/10.3390/mi10110787