
Studies on the Presence of Mycotoxins in Biological Samples: An Overview


Abstract
:1. Introduction
2. Results and Discussion
2.1. Mycotoxins Analysis in Biological Fluids
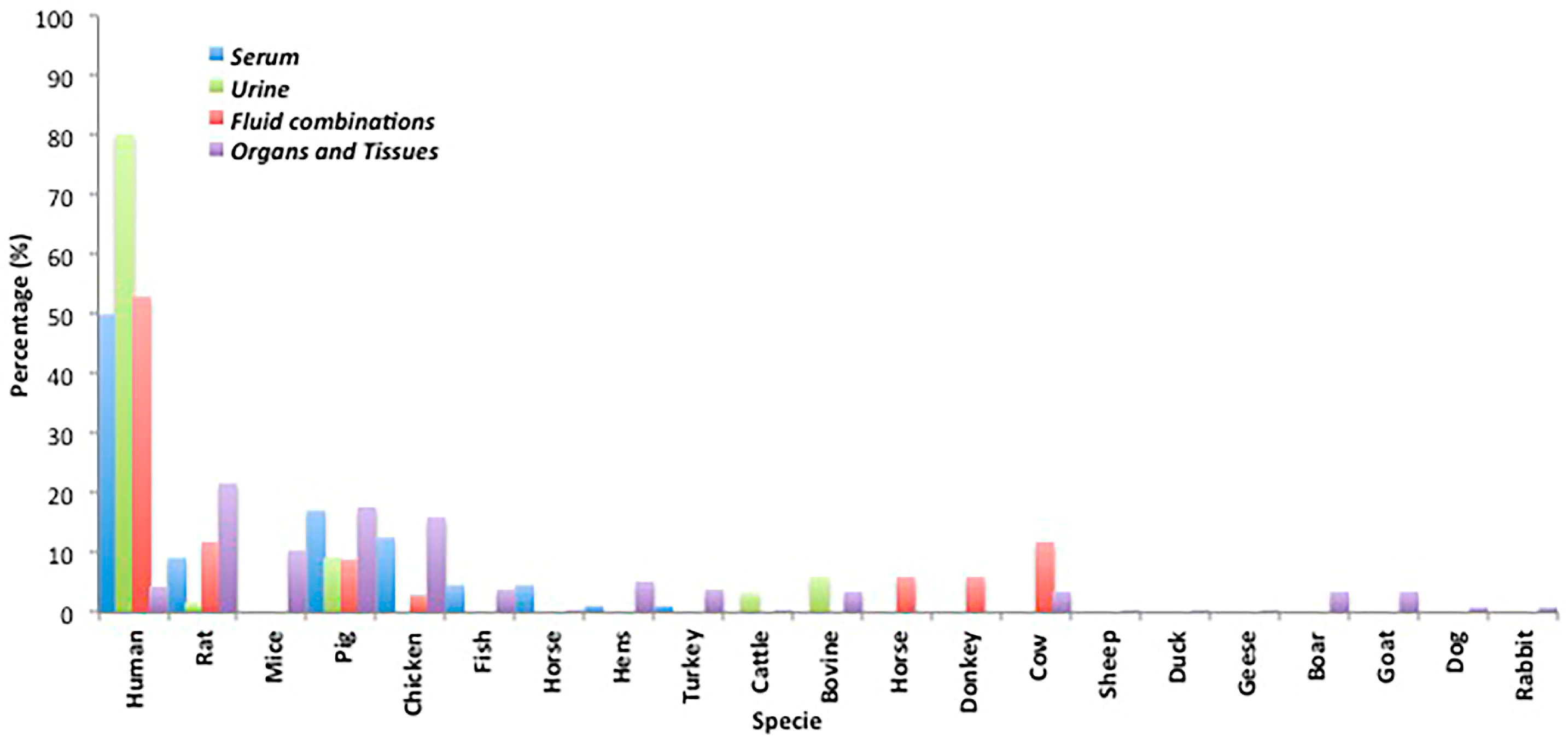
2.1.1. Serum
2.1.2. Urine
2.1.3. Minor Biological Fluids and Fluid Combinations
2.2. Mycotoxin Analysis in Organs and Tissues
2.3. Most Common Methodologies
2.4. Most Studied Mycotoxins
2.5. Biological Sample Origin
2.6. Expected Purposes of Biological Sample Analysis
2.7. Mycotoxin Bioaccumulation Findings
2.7.1. DON and Metabolites
2.7.2. ZON and Metabolites
2.7.3. OTA
2.7.4. ENs and BEA
2.7.5. NIV and FUS-X
2.7.6. T-2 and HT-2
2.7.7. FBs
2.8. Risk Assessment
2.8.1. OTA
2.8.2. DON and Metabolites
2.8.3. CIT
2.8.4. Multi-Mycotoxins
2.8.5. Mycotoxin Binders
3. Conclusions
4. Materials and Methods
Data Resource
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Turner, N.W.; Bramhmbhatt, H.; Szabo-Vezse, M.; Poma, A.; Coker, R.; Piletsky, S.A. Analytical methods for determination of mycotoxins: An update (2009–2014). Anal. Chim. Acta 2015, 9011, 2–33. [Google Scholar] [CrossRef] [PubMed]
- Tsitsigiannis, I.; Antoniou, P.; Tjamos, C. Biological control strategies of mycotoxigenic fungi and associated mycotoxins in Mediterranean basin crops. Phytopathol. Mediterr. 2012, 51, 158–174. [Google Scholar]
- Xu, L.; Zhang, Z.; Zhang, Q.; Li, P. Mycotoxin determination in foods using advanced sensors based on antibodies or aptamers. Toxins 2016, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, J.; Jiang, Y.; Duan, X.; Qu, H.; Yang, B.; Chen, F.; Sivakumar, D. Natural Occurrence, Analysis, and Prevention of Mycotoxins in Fruits and their Processed Products. Crit. Rev. Food Sci. Nutr. 2014, 54, 64–83. [Google Scholar] [CrossRef] [PubMed]
- Zollner, P.; Mayer-Helm, B. Trace mycotoxin analysis in complex biological and food matrices by liquid chromatography-atmospheric pressure ionisation mass spectrometry. J. Chromatogr. A 2006, 1136, 123–169. [Google Scholar] [CrossRef] [PubMed]
- Escrivá, L.; Font, G.; Manyes, L. In vivo toxicity studies of Fusarium mycotoxins in the last decade: A review. Food Chem. Toxicol. 2015, 78, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Guerre, P. Fusariotoxins in Avian Species: Toxicokinetics, Metabolism and Persistence in Tissues. Toxins 2015, 7, 2289–2305. [Google Scholar] [CrossRef] [PubMed]
- Abrunhosa, L.; Morales, H.; Soares, C.; Calado, T.; Vila-Chã, A.S.; Pereira, M.; Venâncio, A. A Review of Mycotoxins in Food and Feed Products in Portugal and Estimation of Probable Daily Intakes. Crit. Rev. Food Sci. Nutr. 2016, 56, 249–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, A.; Susca, A.; Mulé, G.; Logrieco, A.F.; Proctor, R.H. Molecular biodiversity of mycotoxigenic fungi that threaten food safety. Int. J. Food Microbiol. 2013, 167, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Summerell, B.A.; Leslie, J.F. Fifty years of Fusarium: How could nine species have ever been enough? Fungal Divers. 2011, 50, 135–144. [Google Scholar] [CrossRef]
- Ismaiel, A.A.; Papenbrock, J. Mycotoxins: Producing Fungi and Mechanisms of Phytotoxicity. Agriculture 2015, 5, 492–537. [Google Scholar] [CrossRef]
- Soto, J.B.; Ruiz, M.J.; Manyes, L.; Juan-García, A. Blood, breast milk and urine: Potential biomarkers of exposure and estimated daily intake of ochratoxin A: A review. Food Addit. Contam. Part A 2015, 33, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Chen, M.; Ying, Y. Development of Methods for Determination of Aflatoxins. Crit. Rev. Food Sci. Nutr. 2016, 56, 2642–2664. [Google Scholar] [CrossRef] [PubMed]
- López, P.; Venema, D.; de Rijk, T.; de Kok, A.; Scholten, J.M.; Mol, H.G.J.; de Nijs, M. Occurrence of Alternaria toxins in food products in The Netherlands. Food Control 2016, 60, 196–204. [Google Scholar] [CrossRef]
- European Food Safety Authority; Panel, E.; Chain, F. Scientific Opinion on the risks for animal and public health related to the presence of Alternaria toxins in feed and food. EFSA J. 2011, 9, 1–97. [Google Scholar]
- IARC Agents Classified by the IARC Monographs, Volumes 1–115. Available online: http://monographs.iarc.fr/ENG/Classification/List_of_Classifications_Vol1-115.pdf (accessed on 3 February 2017).
- Kwaśniewska, K.; Gadzała-Kopciuch, R.; Cendrowski, K. Analytical Procedure for the Determination of Zearalenone in Environmental and Biological. Crit. Rev. Anal. Chem. 2015, 45, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Schneweis, I.; Meyer, K.; Ritzmann, M.; Hoffmann, P.; Dempfle, L.; Bauer, J. Influence of organically or conventionally produced wheat on health, performance and mycotoxin residues in tissues and bile of growing pigs. Arch. Anim. Nutr. 2005, 59, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Amuzie, C.J.; Harkema, J.R.; Pestka, J.J. Tissue distribution and proinflammatory cytokine induction by the trichothecene deoxynivalenol in the mouse: Comparison of nasal vs. oral exposure. Toxicology 2008, 248, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Danicke, S.; Brezina, U. Kinetics and metabolism of the Fusarium toxin deoxynivalenol in farm animals: Consequences for diagnosis of exposure and intoxication and carry over. Food Chem. Toxicol. 2013, 60, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Minervini, F.; Giannoccaro, A.; Nicassio, M.; Panzarini, G.; Lacalandra, J.M. First Evidence of Placental Transfer of Ochratoxin A in Horses. Toxins 2013, 5, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Britzi, M.; Friedman, S.; Miron, J.; Solomon, R.; Cuneah, O.; Shimshoni, J.A.; Soback, S.; Ashkenazi, R.; Armer, S.; Shlosberg, A. Carry-Over of Aflatoxin B1 to Aflatoxin M1 in High Yielding Israeli Cows in Mid- and Late-Lactation. Toxins 2013, 5, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Tozzi, B.; Liponi, G.B.; Meucci, V.; Casini, L.; Dall’Asta, C.; Intorre, L.; Gatta, D. Aflatoxins M1 and M2 in the milk of donkeys fed with naturally contaminated diet. Dairy Sci. Technol. 2016, 96, 513–523. [Google Scholar] [CrossRef]
- Winkler, J.; Kersten, S.; Valenta, H.; Meyer, U.; Engelhardt, U.H.; Dänicke, S. Development of a multi-toxin method for investigating the carryover of zearalenone, deoxynivalenol and their metabolites into milk of dairy cows. Food Addit. Contam. Part A 2015, 32, 371–380. [Google Scholar]
- Jonsson, M.; Jestoi, M.; Anthoni, M.; Welling, A.; Loivamaa, I.; Hallikainen, V.; Kankainen, M.; Lysøe, E.; Koivisto, P.; Peltonen, K. Fusarium mycotoxin enniatin B: Cytotoxic effects and changes in gene expression profile. Toxicol. In Vitro 2016, 34, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carrasco, Y.; Heilos, D.; Richter, L.; Süssmuth, R.D.; Heffeter, P.; Sulyok, M.; Kenner, L.; Berger, W.; Dornetshuber-Fleiss, R. Mouse tissue distribution and persistence of the food-born fusariotoxins Enniatin B and Beauvericin. Toxicol. Lett. 2016, 247, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Armorini, S.; Al-Qudah, K.M.; Altafini, A.; Zaghini, A.; Roncada, P. Biliary ochratoxin A as a biomarker of ochratoxin exposure in laying hens: An experimental study after administration of contaminated diets. Res. Vet. Sci. 2015, 100, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wu, S.; Yue, Y.; Wang, S.; Wang, Y.; Tao, L.; Tian, H. A high-throughput method for the simultaneous determination of multiple mycotoxins in human and laboratory animal biological fluids and tissues by PLE and HPLC-MS/MS. J. Chromatogr. B 2013, 942–943, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.B.; Font, G.; Mañes, J.; Ferrer, E. Dispersive Liquid-Liquid Microextraction for the Determination of Emerging Fusarium Mycotoxins in Water. Food Anal. Meth. 2016, 9, 856–862. [Google Scholar] [CrossRef]
- Song, S.; Ediage, E.N.; Wu, A.; De Saeger, S. Development and application of salting-out assisted liquid/liquid extraction for multi-mycotoxin biomarkers analysis in pig urine with high performance liquid chromatography/tandem mass spectrometry. J. Chromatogr. A 2013, 1292, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carrasco, Y.; Mañes, J.; Berrada, H.; Font, G. Preliminary Estimation of Deoxynivalenol Excretion through a 24 h Pilot Study. Toxins 2015, 7, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Danicke, S.; Winkler, J. Diagnosis of zearalenone (ZEN) exposure of farm animals and transfer of its residues into edible tissues (carry over). Food Chem. Toxicol. 2015, 84, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Suomela, J.-P.; Jarvinen, R.; Lassila, M. Derivatization. In First Dice Your Dill (Anethum graveolens L.) New Methods and Techniques in Sample Handling; University of Turku FI-20014: Turku, Finland, 2010. [Google Scholar]
- Liu, L.H.; Zhou, X.H.; Shi, H.C. Portable optical aptasensor for rapid detection of mycotoxin with a reversible ligand-grafted biosensing surface. Biosens. Bioelectron. 2015, 72, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Degen, G.H.; Mayer, S.; Blaszkewicz, M. Biomonitoring of ochratoxin A in grain workers. Mycotoxin Res. 2007, 23, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Medina, Á.; Mateo, E.M.; Roig, R.J.; Blanquer, A.; Jiménez, M. Ochratoxin A levels in the plasma of healthy blood donors from Valencia and estimation of exposure degree: Comparison with previous national Spanish data. Food Addit. Contam. Part A 2010, 27, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Rivzi, S.A.H.; Beg, A.E.; Blaszkewicz, M.; Golka, K.; Degen, G.H. Analysis of Ochratoxin a Blood Levels in Bladder Cancer Cases and Healthy Persons from Pakistan. J. Toxicol. Environ. Health Part A 2012, 75, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Dohnal, V.; Dvorák, V.; Malír, F.; Ostry, V.; Roubal, T. A comparison of ELISA and HPLC methods for determination of ochratoxin A in human blood serum in the Czech Republic. Food Chem. Toxicol. 2013, 62, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Koller, G.; Wichmann, G.; Rolle-Kampczyk, U.; Popp, P.; Herbarth, O. Comparison of ELISA and capillary electrophoresis with laser-induced fluorescence detection in the analysis of Ochratoxin A in low volumes of human blood serum. J. Chromatogr. B 2016, 840, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Kruger, C.D.; Cavaglieri, L.R.; Direito, G.M.; Keller, K.M.; Dalcero, A.M.; da Rocha Rosa, C.A. Ochratoxin A in serum of swine from different Brazilian states. J. Vet. Diagn. Investig. 2010, 22, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Hmaissia Khlifaa, K.; Ghalib, R.; Mazigha, C.; Aounia, Z.; Machgoula, S.; Hedhili, A. Ochratoxin A levels in human serum and foods from nephropathy patients in Tunisia: Where are you now? Exp. Toxicol. Pathol. 2012, 64, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Sangare-Tigori, B.; Moukha, S.; Kouadio, J.H.; Dano, D.S.; Betbeder, A.-M.; Achour, A.; Creppy, E.E. Ochratoxin A in human blood in Abidjan, Cote d’Ivoire. Toxicon 2006, 47, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Malir, F.; Ostry, V.; Dofkova, M.; Roubal, T.; Dvorak, V.; Dohnal, V. Ochratoxin A levels in blood serum of Czech women in the first trimester of pregnancy and its correspondence with dietary intake of the mycotoxin contaminant. Biomarkers 2013, 18, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, K.; Vega, M.; Rios, G.; Muñoz, S.; Madariaga, R. Preliminary study of Ochratoxin A in human plasma in agricultural zones of Chile and its relation to food consumption. Food Chem. Toxicol. 2006, 44, 1884–1889. [Google Scholar] [CrossRef] [PubMed]
- Fraeyman, S.; Devreese, M.; Antonissen, G.; De Baere, S.; Rychlik, M.; Croubels, S. Comparative Oral Bioavailability, Toxicokinetics, and Biotransformation of Enniatin B1 and Enniatin B in Broiler Chickens. J. Agric. Food Chem. 2016, 64, 7259–7264. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; Osselaere, A.; Goossens, J.; Vandenbroucke, V.; De Baere, S.; Eeckhout, M.; De Backer, P.; Croubels, S. New bolus models for in vivo efficacy testing of mycotoxin-detoxifying agents in relation to EFSA guidelines, assessed using deoxynivalenol in broiler chickens. Food Addit. Contam. Part A 2012, 29, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; Girgis, G.N.; Tran, S.-T.; De Baere, S.; De Backer, P.; Croubels, S.; Smith, T.K. The effects of feed-borne Fusarium mycotoxins and glucomannan in turkey poults based on specific and non-specific parameters. Food Chem. Toxicol. 2014, 63, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.S.; Hong, S.H.; Hwang, S.W.; Kim, H.J.; Lee, J.B.; Yoon, H.-S.; Kim, D.J.; Yoo, S.D. Determination of zearalenone by liquid chromatography/tandem mass spectrometry and application to a pharmacokinetic study. Biomed. Chromatogr. 2009, 23, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.S.; Hong, S.H.; Kim, H.J.; Yoon, H.-S.; Kim, D.J.; Hwang, S.W.; Lee, J.B.; Yoo, S.D. Development of a Sensitive LC Assay with Fluorescence Detection for the Determination of Zearalenone in Rat Serum. Chromatographia 2009, 69, 295–299. [Google Scholar] [CrossRef]
- Saad-Hussein, A.; Taha, M.M.; Fadl, N.N.; Awad, A.-H.; Mahdy-Abdallah, H.; Moubarz, G.; Aziz, H.; El-Shamy, K.A. Effects of airborne Aspergillus on serum aflatoxin B1 and liver enzymes in workers handling wheat flour. Hum. Exp. Toxicol. 2016, 35, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; De Baere, S.; De Backer, P.; Croubels, S. Quantitative determination of several toxicological important mycotoxins in pig plasma using multi-mycotoxin and analyte-specific high performance liquid chromatography–tandem mass spectrometric methods. J. Chromatogr. A 2012, 1257, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; De Baere, S.; De Backer, P.; Croubels, S. Quantitative determination of the Fusarium mycotoxins beauvericin, enniatin A, A1, B and B1 in pig plasma using high performance liquid chromatography–tandem mass spectrometry. Talanta 2013, 106, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; Broekaert, N.; De Mil, T.; Fraeyman, S.; De Backer, P.; Croubels, S. Pilot toxicokinetic study and absolute oral bioavailability of the Fusarium mycotoxin enniatin B1 in pigs. Food Chem. Toxicol 2014, 63, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Devreese, M.; Antonissen, G.; De Backer, P.; Croubels, S. Efficacy of Active Carbon towards the Absorption of Deoxynivalenol in Pigs. Toxins 2014, 6, 2998–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devreese, M.; Antonissen, G.; Broekaert, N.; De Baere, S.; Vanhaecke, L.; De Backer, P.; Croubels, S. Comparative Toxicokinetics, Absolute Oral Bioavailability, and Biotransformation of Zearalenone in Different Poultry Species. J. Agric. Food Chem. 2015, 63, 5092–5098. [Google Scholar] [CrossRef] [PubMed]
- Broekaert, N.; Devreese, M.; De Mil, T.; Fraeyman, S.; De Baere, S.; De Saeger, S.; De Backer, P.; Croubels, S. Development and validation of an LC-MS/MS method for the toxicokinetic study of deoxynivalenol and its acetylated derivatives in chicken and pig plasma. J. Chromatogr. B 2014, 971, 43–51. [Google Scholar] [CrossRef] [PubMed]
- De Baere, S.; Osselaere, A.; Devreese, M.; Vanhaecke, L.; De Backer, P.; Croubels, S. Development of a liquid-chromatography tandem mass spectrometry and ultra-high-performance liquid chromatography high-resolution mass spectrometry method for the quantitative determination of zearalenone and its major metabolites in chicken and pig plasma. Anal. Chim. Acta 2012, 756, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Blaszkewicz, M.; Manirujjaman, M.; Perveen, R.; Al Nahid, A.; Mahmood, S.; Rahman, M.; Hossain, K.; Degen, G.H. Biomonitoring of ochratoxin A in blood plasma and exposure assessment of adult students in Bangladesh. Mol. Nutr. Food Res. 2014, 58, 2219–2225. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, J.; Font, G.; Mañes, J.; Ferrer, E. Multimycotoxin analysis in water and fish plasma by liquid chromatography-tandem mass spectrometry. Chemosphere 2016, 145, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pena, A.; Seifrtova, M. Estimation of ochratoxin A in portuguese population: New data on the occurrence in human urine by high performance liquid chromatography with fluorescence detection. Food Chem. Toxicol. 2006, 44, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Manique, R.; Pena, A.; Lino, C.M.; Moltó, J.C.; Mañes, J. Ochratoxin A in the morning and afternoon portions of urine from Coimbra and Valencian populations. Toxicon 2008, 51, 1281–1287. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Muñoz, K.; Degen, G.H. Ochratoxin A and its metabolites in urines of German adults—An assessment of variables in biomarker analysis. Toxicol. Lett. 2017, 275, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Vatinno, R.; Vuckovic, D.; Zambonin, C.G.; Pawliszyn, J. Automated high-throughput method using solid-phase microextraction-liquid chromatography-tandem mass spectrometry for the determination of ochratoxin A in human urine. J. Chromatogr. A 2008, 1201, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, K.; Cramer, B.; Dopstadt, J.; Humpf, H.U.; Degen, G.H. Evidence of ochratoxin A conjugates in urine samples from infants and adults. Mycotoxin Res. 2017, 33, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Fushimi, Y.; Takagi, M.; Uno, S.; Kokushi, E.; Nakamura, M.; Hasunuma, H.; Shinya, U.; Deguchi, E.; Fink-Gremmels, J. Measurement of Sterigmatocystin Concentrations in Urine for Monitoring the Contamination of Cattle Feed. Toxins 2014, 6, 3117–3128. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Sulyok, M.; Berthiller, F.; Schuhmacher, R.; Fruhmann, P.; Hametner, C.; Adam, G.; Fröhlich, J.; Krska, R. Direct quantification of deoxynivalenol glucuronide in human urine as biomarker of exposure to the Fusarium mycotoxin deoxynivalenol. Anal. Bioanal. Chem. 2011, 401, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Egner, P.A.; Groopman, J.D.; Wang, J.-S.; Kensler, T.W.; Friesen, M.D. Quantification of Aflatoxin-B1-N7-Guanine in Human Urine by High-Performance Liquid Chromatography and Isotope Dilution Tandem Mass Spectrometry. Chem. Res. Toxicol. 2006, 19, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Gerding, J.; Cramer, B.; Humpf, H. Determination of mycotoxin exposure in Germany using an LC-MS/MS multibiomarker approach. Mol. Nutr. Food Res. 2014, 58, 2358–2368. [Google Scholar] [CrossRef] [PubMed]
- Gerding, J.; Ali, N.; Schwartzbord, J.; Cramer, B.; Brown, D.L.; Degen, G.H.; Humpf, H. A comparative study of the human urinary mycotoxin excretion patterns in Bangladesh, Germany, and Haiti using a rapid and sensitive LC-MS/MS approach. Mycotoxin Res. 2015, 31, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Ezekiel, C.N.; Warth, B.; Ogara, I.M.; Abia, W.A.; Ezekiel, V.C.; Atehnkeng, J.; Sulyok, M.; Turner, P.C.; Tayo, G.O.; Krska, R.; et al. Mycotoxin exposure in rural residents in northern Nigeria: A pilot study using multi-urinary biomarkers. Environ. Int. 2014, 66, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Sulyok, M.; Fruhmann, P.; Mikula, H.; Berthiller, F.; Schuhmacher, R.; Hametner, C.; Abia, W.A.; Adam, G.; Fröhlich, G.; et al. Development and validation of a rapid multi-biomarker liquid chromatography/tandem mass spectrometry method to assess human exposure to mycotoxins. Rapid Commun. Mass Spectrom. 2012, 26, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Sulyok, M.; Fruhmann, P.; Berthiller, F.; Schuhmacher, R.; Hametner, C.; Adam, G.; Fröhlich, J.; Krska, R. Assessment of human deoxynivalenol exposure using an LC-MS/MS based biomarker method. Toxicol. Lett. 2012, 211, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Heyndrickx, E.; Sioen, I.; Bellemans, M.; De Maeyer, M.; Callebaut, A.; De Henauw, S.; De Saeger, S. Assessment of mycotoxin exposure in the Belgian population using biomarkers: Aim, design and methods of the BIOMYCO study. Food Addit. Contam. Part A 2014, 31, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Heyndrickx, E.; Sioen, I.; Huybrechts, B.; Callebaut, A.; De Henauw, S.; De Saeger, S. Human biomonitoring of multiple mycotoxins in the Belgian population: Results of the BIOMYCO study. Environ. Int. 2015, 84, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Andrés, F.; Zougagh, M.; Casta, G.; Ríos, A. Determination of zearalenone and its metabolites in urine samples by liquid chromatography with electrochemical detection using a carbon nanotube-modified electrode. J. Chromatogr. A 2008, 121, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, V.M.T.; Solfrizzo, M.; De Girolamo, A.; Chulze, S.N.; Torres, A.M.; Visconti, A. LC-MS/MS characterization of the urinary excretion profile of the mycotoxin deoxynivalenol in human and rat. J. Chromatogr. B 2011, 879, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzo, M.; Gambacorta, L.; Visconti, A. Assessment of Multi-Mycotoxin Exposure in Southern Italy by Urinary Multi-Biomarker Determination. Toxins 2014, 6, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Blokland, M.H.; Sterk, S.S.; Stephany, R.W.; Launay, F.M.; Kennedy, D.G.; van Ginkel, L.A. Determination of resorcylic acid lactones in biological samples by GC-MS. Discrimination between illegal use and contamination with Fusarium toxins. Anal. Bioanal. Chem. 2006, 384, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Ediage, E.N.; Di Mavungua, J.D.; Song, S.; Wu, A.; Van Peteghem, C.; De Saeger, S. A direct assessment of mycotoxin biomarkers in human urine samples by liquid chromatography tandem mass spectrometry. Anal. Chim. Acta 2012, 741, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solfrizzo, M.; Gambacorta, L.; Lattanzio, V.M.T.; Powers, S.; Visconti, A. Simultaneous LC-MS/MS determination of aflatoxin M1, ochratoxin A, deoxynivalenol, de-epoxydeoxynivalenol, α and β-zearalenols and fumonisin B1 in urine as a multi-biomarker method to assess exposure to mycotoxins. Anal. Bioanal. Chem. 2011, 401, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Gambacorta, S.; Solfrizzo, H.; Visconti, A.; Powers, S.; Cossalter, A.M.; Pinton, P.; Oswald, I.P. Validation study on urinary biomarkers of exposure for aflatoxin B1, ochratoxin A, fumonisin B1, deoxynivalenol and zearalenone in piglets. World Mycotoxin J. 2013, 6, 299–308. [Google Scholar] [CrossRef]
- Ahn, J.; Kim, D.; Kim, H.; Jahng, K.-Y. Quantitative determination of mycotoxins in urine by LC-MS/MS. Food Addit. Contam. Part A 2010, 27, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Rubert, J.; Soriano, J.M.; Mañes, J.; Soler, C. Rapid mycotoxin analysis in human urine: A pilot study. Food Chem. Toxicol. 2011, 49, 2299–2304. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.C.; Ji, B.T.; Shu, X.O.; Zheng, W.; Chow, W.-H.; Gao, Y.T.; Hardie, L.J. A biomarker survey of urinary deoxynivalenol in China: The Shanghai Women’s Health Study. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2011, 28, 1220–1223. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Blaszkewicz, M.; Al Nahid, A.; Rahman, M.; Degen, G.H. Deoxynivalenol Exposure Assessment for Pregnant Women in Bangladesh. Toxins 2015, 7, 3845–3857. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Blaszkewicz, M.; Degen, G.H. Assessment of deoxynivalenol exposure among Bangladeshi and German adults by a biomarker-based approach. Toxicol. Lett. 2016, 258, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Blaszkewicz, M.; Degen, G.H. Occurrence of the mycotoxin citrinin and its metabolite dihydrocitrinone in urines of German adults. Arch. Toxicol. 2015, 89, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Blaszkewicz, M.; Mohanto, N.C.; Rahman, M.; Alim, A.; Hossain, K.; Degen, G.H. First results on citrinin biomarkers in urines from rural and urban cohorts in Bangladesh. Mycotoxin Res. 2015, 31, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.J.G.; Pena, A.; Lino, C.M.; Fernández, M.F.; Mañes, J. Fumonisins determination in urine by LC-MS-MS. Anal. Bioanal. Chem. 2010, 396, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Dusi, G.; Bozzoni, E.; Assini, W.; Tognoli, N.; Gasparini, M.; Ferretti, E. Confirmatory method for the determination of resorcylic acid lactones in urine sample using immunoaffinity cleanup and liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2009, 637, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Rejczak, T.; Tuzimski, T. A review of recent developments and trends in the QuEChERS sample preparation approach. Open Chem. 2015, 13, 980–1010. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Moltó, J.C.; Mañes, J.; Berrada, H. Development of a GC–MS/MS strategy to determine 15 mycotoxins and metabolites in human urine. Talanta 2014, 128, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carrasco, Y.; Moltó, J.C.; Mañes, J.; Berrada, H. Exposure assessment approach through mycotoxin/creatinine ratio evaluation in urine by GC-MS/MS. Food Chem. Toxicol. 2014, 72, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carrasco, Y.; Moltó, J.C.; Mañes, J.; Berrada, H. Development of microextraction techniques in combination with GC-MS/MS for the determination of mycotoxins and metabolites in human urine. J. Sep. Sci. 2017, 40, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, K.; Blaszkewicz, M.; Degen, G.H. Simultaneous analysis of ochratoxin A and its major metabolite ochratoxin alpha in plasma and urine for an advanced biomonitoring of the mycotoxin. J. Chromatogr. B 2010, 878, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Blaszkewicz, M.; Manirujjaman, M.; Degen, G.H. Biomonitoring of concurrent exposure to ochratoxin A and citrinin in pregnant women in Bangladesh. Mycotoxin Res. 2016, 32, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Brezina, U.; Rempe, I.; Kersten, S.; Valenta, H.; Humpf, H.-U.; Dänicke, S. Diagnosis of intoxications of piglets fed with Fusarium toxin-contaminated maize by the analysis of mycotoxin residues in serum, liquor and urine with LC-MS/MS. Arch. Anim. Nutr. 2014, 68, 425–447. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.B.; Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; Samperi, R.; Ventura, S.; Laganà, A. Development of a Rapid LC-MS/MS Method for the Determination of Emerging Fusarium mycotoxins Enniatins and Beauvericin in Human Biological Fluids. Toxins 2015, 7, 3554–3571. [Google Scholar] [CrossRef] [PubMed]
- Blaszkewicz, M.; Muñoz, K.; Degen, G.H. Methods for analysis of citrinin in human blood and urine. Arch. Toxicol. 2013, 87, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Escrivá, L.; Font, G.; Manyes, L. Quantitation of enniatins in biological samples of Wistar rats after oral administration by LC-MS/MS. Toxicol. Mech. Methods 2015, 25, 552–558. [Google Scholar] [PubMed]
- Juan, C.; Manyes, L.; Font, G.; Juan-Garcia, A. Evaluation of immunologic effect of Enniatin A and quantitative determination in feces, urine and serum on treated Wistar rats. Toxicon 2014, 87, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Songsermsakul, P.; Sontag, G.; Cichna-Markl, M.; Zentek, J.; Razzazi-Fazeli, E. Determination of zearalenone and its metabolites in urine, plasma and faeces of horses by HPLC-APCI-MS. J. Chromatogr. B 2006, 843, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Schwartz-Zimmermann, H.E.; Fruhmann, P.; Dänicke, S.; Wiesenberger, G.; Caha, S.; Weber, J.; Berthiller, F. Metabolism of Deoxynivalenol and Deepoxy-Deoxynivalenol in Broiler Chickens, Pullets, Roosters and Turkeys. Toxins 2015, 7, 4706–4729. [Google Scholar] [CrossRef] [PubMed]
- Warth, B.; Sulyok, M.; Krska, R. LC-MS/MS-based multibiomarker approaches for the assessment of human exposure to mycotoxins. Anal. Bioanal. Chem. 2013, 405, 5687–5695. [Google Scholar] [CrossRef] [PubMed]
- Huybrechts, B.; Martins, J.C.; Debongnie, P.; Uhlig, S.; Callebaut, A. Fast and sensitive LC-MS/MS method measuring human mycotoxin exposure using biomarkers in urine. Arch. Toxicol. 2015, 89, 1993–2005. [Google Scholar] [CrossRef] [PubMed]
- Gürbay, A.; Girgin, G.; Sabuncuog, S.A.Ş.; Şahin, G.; Yurdakök, M.; Yig, Ş.; Tekinalp, G. Ochratoxin A: Is it present in breast milk samples obtained from mothers from Ankara, Turkey? J. Appl. Toxicol. 2009, 30, 329–333. [Google Scholar] [CrossRef]
- Afshar, P.; Shokrzadeh, M.; Kalhori, S.; Babaee, Z.; Saravi, S.S.S. Occurrence of Ochratoxin A and A flatoxin M1 in human breast milk in Sari, Iran. Food Control 2013, 31, 525–529. [Google Scholar] [CrossRef]
- Camel, V.; Ouethrani, M.; Coudray, C.; Philippe, C.; Rabot, S. Semi-automated solid-phase extraction method for studying the biodegradation of ochratoxin A by human intestinal microbiota. J. Chromatogr. B 2012, 893–894, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Andrade, P.D.; Gomes da Silva, J.L.; Caldas, E.D. Simultaneous analysis of aflatoxins B1, B2, G1, G2, M1 and ochratoxinA in breast milk by high-performance liquidchromatography/fluorescence after liquid-liquid extraction with lowtemperature purification (LLE-LTP). J. Chromatogr. A 2013, 1304, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Rubert, J.; León, N.; Sáez, C.; Martins, C.P.B.; Godula, M.; Yusà, V.; Mañes, J.; Soriano, J.M.; Soler, C. Evaluation of mycotoxins and their metabolites in human breast milk using liquid chromatography coupled to high resolution mass spectrometry. Anal. Chim. Acta 2014, 820, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Massart, F.; Micillo, F.; Rivezzi, G.; Perrone, L.; Baggiani, A.; Miccoli, M.; Meucci, V. Zearalenone screening of human breast milk from the Naples area. Toxicol. Environ. Chem. 2016, 98, 128–136. [Google Scholar] [CrossRef]
- De Baere, S.; Goossens, J.; Osselaere, A.; Devreese, M.; Vandenbroucke, V.; De Backer, P.; Croubels, S. Quantitative determination of T-2 toxin, HT-2 toxin, deoxynivalenol and deepoxy-deoxynivalenol in animal body fluids using LC-MS/MS detection. J. Chromatogr. B 2011, 879, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Osselaere, A.; Devreese, M.; Goossens, J.; Vandenbroucke, V.; De Baere, S.; De Backer, P.; Croubels, S. Toxicokinetic study and absolute oral bioavailability of deoxynivalenol, T-2 toxin and zearalenone in broiler chickens. Food Chem. Toxicol. 2013, 51, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.G.; Bolton, V.E.; Guilford, F.T.; Straus, D.C. Mycotoxin Detection in Human Samples from Patients Exposed to Environmental Molds. Int. J. Mol. Sci. 2009, 10, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Taevernier, L.; Bracke, N.; Veryser, L.; Wynendaele, E.; Gevaert, B.; Peremans, K.; De Spiegeleer, B. Blood-brain barrier transport kinetics of the cyclic depsipeptide mycotoxins beauvericin and enniatins. Toxicol. Lett. 2016, 258, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Veršilovskis, A.; Geys, J.; Huybrechts, B.; Goossens, E.; De Saeger, S.; Callebaut, A. Simultaneous determination of masked forms of deoxynivalenol and zearalenone after oral dosing in rats by LC-MS/MS. World Mycotoxin J. 2012, 5, 303–318. [Google Scholar] [CrossRef]
- Corcuera, L.; Ibáñez-Vea, M.; Vettorazzi, A.; González-Peñas, E.; López de Cerain, A. Validation of a UHPLC-FLD analytical method for the simultaneous quantification of aflatoxin B1 and ochratoxin a in rat plasma, liver and kidney. J. Chromatogr. B 2011, 879, 2733–2740. [Google Scholar] [CrossRef] [PubMed]
- Manyes, L.; Escriva, L.; Belen Serrano, A.; Rodriguez-Carrasco, Y.; Tolosa, J.; Meca, G.; Font, G. A preliminary study in Wistar rats with enniatin A contaminated feed. Toxicol. Mech. Methods 2014, 24, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, Z.; Wu, A.; Deng, Y.; Zhou, Z. Determination of trichothecenes A (T-2 toxin, HT-2 toxin, and diacetoxyscirpenol) in the tissues of broilers using liquid chromatography coupled to tandem mass spectrometry. J. Chromatogr. B 2013, 942–943, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Milicevic, D.; Juric, V.; Stefanovic, S.; Baltic, T.; Jankovic, S. Evaluation and Validation of Two Chromatographic Methods (HPLC-Fluorescence and LC-MS/MS) for the Determination and Confirmation of Ochratoxin A in Pig Tissues. Arch. Environ. Contam. Toxicol. 2010, 58, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Vettorazzi, A.; Gonzalez-Peñas, E.; Arbillagaa, L.; Corcuera, L.-A.; López de Ceraina, A. Simple high-performance liquid chromatography-fluorescence detection method for plasma, kidney and liver of rat as a tool for toxicology studies. J. Chromatogr. A 2008, 1215, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.S.; Hong, S.H.; Bulitta, J.B.; Hwang, S.W.; Kim, H.J.; Lee, J.B.; Yang, S.D.; Kim, J.E.; Yoon, H.S.; Kim, D.J.; et al. Disposition, Oral Bioavailability, and Tissue Distribution of Zearalenone in Rats at Various Dose Levels Disposition. J. Toxicol. Environ. Health Part A 2009, 72, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Chandratre, G.A.; Telang, A.G.; Badgujar, P.C.; Raut, S.S.; Sharma, A.K. Toxicopathological Alterations Induced by High Dose Dietary T-2 Mycotoxin and its Residue Detection in Wistar Rats. Arch. Environ. Contam. Toxicol. 2014, 67, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Saengtienchai, T.; Poapolathep, S.; Isariyodom, S.; Ikenaka, Y.; Ishizuka, M.; Poapolathep, A. Toxicokinetics and tissue depletion of Fusarenon-X and its metabolite nivalenol in piglets. Food Chem. Toxicol. 2014, 66, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Kongkapan, J.; Giorgi, M.; Poapolathep, S.; Isariyodom, S.; Poapolathep, A. Toxicokinetics and tissue distribution of nivalenol in broiler chickens. Toxicon 2016, 111, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, G.; Zhao, H.; Zheng, J.; Hu, F.; Fang, B. Liquid chromatography-tandem mass spectrometry method for toxicokinetics, tissue distribution, and excretion studies of T-2 toxin and its major metabolites in pigs. J. Chromatogr. B 2014, 958, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Jestoi, M.; Rokka, M.; Peltonen, K. An integrated sample preparation to determine coccidiostats and emerging Fusarium-mycotoxins in various poultry tissues with LC-MS/MS. Mol. Nutr. Food Res. 2007, 51, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, J.; Font, G.; Mañes, J.; Ferrer, E. Natural Occurrence of Emerging Fusarium Mycotoxins in Feed and Fish from Aquaculture. J. Agric. Food Chem. 2014, 62, 12462–12470. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, Y.; Beier, R.C.; Zhang, H.; De Ruyck, K.; Sun, F.; Cao, X.; Shen, J.; Zhang, D.; Wang, Z. Simultaneous Determination of Type A and B Trichothecenes and Their Main Metabolites in Food Animal Tissues by Ultraperformance Liquid Chromatography Coupled with Triple-Quadrupole Mass Spectrometry. J. Agric. Food Chem. 2015, 63, 8592–8600. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, G.; Guo, C.; Zhang, Y.; Zhang, Y.; Zheng, J.; Yang, H.; Yang, D.; He, L.; Zeng, Z.; et al. Simultaneous determination of major type-B trichothecenes and the de-epoxy metabolite of deoxynivalenol in chicken tissues by HPLC-MS/MS. J. Sep. Sci. 2014, 37, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Haiyang, J.; Wenjun, W.; Jinghui, Z.; Xiaoqi, T.; Jiancheng, L.; Xi, X.; Kai, W.; Fei, X.; Zhaopeng, W.; Min, C.; et al. Determination of zeranol and its metabolites in bovine muscle and liver by a chemiluminescence enzyme immunoassay: Compared to an ultraperformance liquid chromatography tandem mass spectroscopy method. Luminescence 2014, 29, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, G.; Zironi, E.; Ceccolini, A.; Matera, R.; Paolo, G.; Piva, A. Simple method for the simultaneous isolation and determination of fumonisin B1 and its metabolite aminopentol-1 in swine liver by liquid chromatography-fluorescence detection. J. Chromatogr. B 2005, 819, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Katsunuma, Y.; Nunokawa, M.; Minato, H.; Yonemochi, C. Influence of repeated ochratoxin A ingestion on milk production and its carry-over into the milk, blood and tissues of lactating cows. Anim. Sci. J. 2016, 87, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewska-Dmytrow, H.; Żmudzki, J.; Burek, O.; Pietruszka, K. Official control of ochratoxin A in food of animal origin in Poland between 2003 and 2012. J. Nat. Vet. Res. Inst. Pulawy 2013, 57, 519–523. [Google Scholar]
- Dong, M.; He, X.J.; Tulayakul, P.; Li, J.-Y.; Dong, K.-S.; Manabe, N.; Nakayama, H.; Kumagai, S. The toxic effects and fate of intravenously administered zearalenone in goats. Toxicon 2010, 55, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Gajecka, M.; Sławuta, P.; Nicpon, J.; Kołacz, R.; Kiełbowicz, Z.; Zielonka, L.; Dąbrowski, M.; Szweda, W.; Gajecki, M.; Nicpon, J. Zearalenone and its metabolites in the tissues of female wild boars exposed per os to mycotoxins. Toxicon 2016, 114, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Danicke, S.; Beyer, M.; Breves, G.; Valenta, H.; Humpf, H.-U. Effects of oral exposure of pigs to deoxynivalenol (DON) sulfonate (DONS) as the non-toxic derivative of DON on tissue residues of DON and de-epoxy-DON and on DONS blood levels. Food Addit. Contam. Part A 2010, 27, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, D.; Bailly, J.; Skiba, F.; Grosjean, F.; Guerre, P. Toxicokinetics of fumonisin B1 in turkey poults and tissue persistence after exposure to a diet containing the maximum European tolerance for fumonisins in avian feeds. Food Chem. Toxicol. 2008, 46, 3213–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestka, J.J.; Islam, Z.; Amuzie, C.J. Immunochemical assessment of deoxynivalenol tissue distribution following oral exposure in the mouse. Toxicol. Lett. 2008, 178, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Mally, A.; Solfrizzo, M.; Degen, G.H. Biomonitoring of the mycotoxin Zearalenone: Current state-of-the art and application to human exposure assessment. Arch. Toxicol. 2016, 90, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Ediage, E.N.; Diana, J.; Mavungu, D.; Song, S.; Sioen, I.; De Saeger, S. Multimycotoxin analysis in urines to assess infant exposure: A case study in Cameroon. Environ. Int. 2013, 57–58, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zollner, P.; Jodlbauer, J.; Kleinova, M.; Kahlbacher, H.; Kuhn, T.; Hochsteiner, W.; Lindner, W. Concentration Levels of Zearalenone and Its Metabolites in Urine, Muscle Tissue, and Liver Samples of Pigs Fed with Mycotoxin-Contaminated Oats. J. Agric. Food Chem. 2002, 50, 2494–2501. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority. Opinion of the scientific panel on contaminants in the food chain on a request from the commission related to ochratoxin A in food. EFSA J. 2006, 365, 1–56. [Google Scholar]
- European Commission. Assessment of Dietary Intake of Ochratoxin A by the Population of European Union Members States. Directorate General-Health and Consumer Protection. Report on Tasks for Scientific Cooperation. Report of Experts Participating in Task 3.2.7; European Commission: Brussels, Belgium; Luxembourg, 2002; pp. 18–19. [Google Scholar]
- European Food Safety Authority. Panel on Contaminants in the Food Chain. Scientific opinion on the risks for public and animal health related to the presence of citrinin in food and feed. EFSA J. 2012, 10, 2605. [Google Scholar]
- Scientific Committee on Food (SCF). Updated Opinion of the Scientific Committee on Food on Fumonisin B1, B2 and B3: SCF/CS/CNTM/MYC/28 Final; SCF: Brussel, Belgium, 2003. [Google Scholar]
- Warth, B.; Braun, D.; Ezekiel, C.N.; Turner, P.C.; Degen, G.H.; Marko, D. Biomonitoring of Mycotoxins in Human Breast Milk: Current State and Future Perspectives. Chem. Res. Toxicol. 2016, 29, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Kolossova, A.; Stroka, J.; Breidbach, A.; Kroeger, K.; Ambrosio, M.; Bouten, K.; Ulberth, F. Evaluation of the Effect of Mycotoxin Binders in Animal Feed on the Analytical Performance of Standardised Methods for the Determination of Mycotoxins in Feed; JRC Scientific and Technical Reports; EUR 23997 EN; Joint Research Centre Institute for Reference Materials and Measurements: Geel, Belgium, 2009; pp. 1–49. [Google Scholar]
- Jacela, J.Y.; De Rouchey, J.M.; Tokach, M.D.; Goodband, R.D.; Nelssen, J.L.; Renter, D.G.; Dritz, S.S. Feed additives for swine: Fact sheets-flavors and mold inhibitors, mycotoxin binders, and antioxidants. J. Swine Health Prod. 2010, 18, 27–32. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Species | Volume (μL) | Mycotoxin | Extraction Procedure | Detection Technique | LOD (μg/L) | LOQ (μg/L) | Reference |
|---|---|---|---|---|---|---|---|
| Single mycotoxin | |||||||
| Human | 50 | OTA | LLE: CH2Cl2 | ELISA vs. CE-LIF (CE/laser-induced FD) | 0.5 | - | [39] |
| Human | - | OTA | IAC | HPLC-FD | - | - | [42] |
| Human | - | OTA | SPE: Sep-Pak RP-18. IAC: Ochraprep | HPLC-FD | 0.1 | 0.4 | [44] |
| Human | - | OTA | LLE: CHCl3/HCl | HPLC-FD | 0.05 | - | [35] |
| Human | 2000 | OTA | LLE: CHCl3 | HPLC-FD LC-ESI-MS/MS | 0.01 | 0.07 | [36] |
| Human | 6000 | OTA | SPE: C18 | HPLC-FD | 0.1 | 0.2 | [41] |
| Human | 1000 | OTA | LLE: CHCl3, SPE | HPLC-FD | 0.05 | - | [37] |
| Human | 2000–3000 | OTA | LLE: CHCl3, IAC: Ochraprep | ELISA and HPLC-FD | - | 0.050 | [38] |
| Human | - | OTA | IAC: Ochraprep | HPLC-FD | - | 0.1 | [43] |
| Human | - | AFB1 | IAC: Easi-Extract Aflatoxin | ELISA | - | - | [50] |
| Rat | 100 | ZON | LLE: TBME | LC-MS/MS | - | 0.5 | [48] |
| Rat | 100 | ZON | LLE: TBME | HPLC-FD | - | 10 | [49] |
| Swine | 800 | OTA | LLE: CH2Cl2 | HPLC | 0.1 | - | [40] |
| Chicken | 250 | DON | LLE: ACN | LC-MS/MS | 0.1–0.2 | 1 | [46,47] |
| Horse | 2000 | OTA | LLE: CH2Cl2, IAC: Ochratest | ELISA, HPLC-FD | 0.015 | - | [21] |
| Multi-mycotoxin | |||||||
| Human | 500 | OTA, OTα | LLE: CHCl3/isopropanol | HPLC-FD | 0.05 | 0.1 | [58] |
| Chicken | 20–250 | EN B, EN B1 | LLE: ACN | LC/MS/MS, UHPLC-HRMS | 0.000091–0.00017 | 0.025 | [45] |
| Pig | 250 | EN A, A1, B, B1 | LLE: ACN | LC-MS/MS | 0.01–0.001 | 0.1–0.2 | [52,53] |
| Pig | 250 | DON, T-2, HT-2, OTA, FB1, AFB1, ZON, ZAN, α-ZOL, β-ZOL, α-ZAL, β-ZAL, DOM-1 | LLE: ACN | LC-MS/MS | 0.01–0.52 | 0.5–10 | [51,54] |
| Chicken, pig, laying hens, turkey poults | 250 | ZON, α-ZOL, β-ZOL, ZAN, α-ZAL, β-ZAL | LLE: ACN | LC–MS/MS (U)HPLC–HR-MS | 0.004–0.07 | 0.2–5 | [55,57] |
| Chicken, Pig | 250 | DON, 3-ADON, 15-ADON, DOM-1 | LLE: ACN | LC–MS/MS | 0.01–0.7 | 0.1–2 | [56] |
| Fish | 250 | AFB1, AFB2, AFG1, AFG2, OTA, FUS-X, STG, FB1, FB2, FB3, BEA, EN A, EN A1, EN B, EN B1 | DLLME: ACN/EtOAc | LC/MS/MS | 0.1–12.0 | 1.5–17.0 | [59] |
| Species | Sample Volume | Mycotoxins | Extraction Procedure | Detection Technique | LOD (μg/L) | LOQ (μg/L) | Reference |
|---|---|---|---|---|---|---|---|
| Single mycotoxin | |||||||
| Human | 10 mL | OTA | IAC: OchraTest | HPLC-FD | - | 0.02 | [60] |
| Human | 1 mL (0.5 mL) | OTA | SPME | HPLC-ESI-MS/MS | 0.3 | 0.7 | [63] |
| Human | 10 mL | OTA | IAC: OchraTest | HPLC-FD | - | 0.007 | [61] |
| Human | 5 mL | OTA, OTα | IAC | HPLC-FD | 0.05 | 0.1 | [62] |
| Human | 20 mL | AFB1-N7-Gua | SPE: MCX, IAC: Bond elute LRC, SPE: C18 | HPLC-ESI-MS/MS | - | - | [67] |
| Human | 0.5 mL | DON-GlcA | Dilute-shoot: ACN/H2O | LC-MS/MS | 3–6 | 10–20 | [66] |
| Cattle | 0.5 mL | STG | SPE: C18 | LC-MS/MS | - | - | [65] |
| Multi-mycotoxin | |||||||
| Human | 1 mL | DON, DOM-1 | IAC: Wide Bore DON | LC-MS/MS | 0.5 | - | [84] |
| Human | 0.1 mL | DON, DON-GlcAs | Dilute-shoot: ACN/H2O | LC-MS/MS | 4–10 | 13–33 | [72] |
| Human Rat | 0.4 | DON, DOM-1, DOM-1-G, DON-G1, DON-G2 | SPE: Strata-X | HPLC-APCI-MS/MS | 1–2 | 3–6 | [76] |
| Human | 10 mL | FB1, FB2 | PBS, IAC | HPLC-ESI-MS/MS | 5 | 10 | [89] |
| Human Swine Bovine | 10 mL | ZON, α-ZOL, β-ZOL, ZAN, α-ZAL, β-ZAL | SPE: C18 | HPLC-EC | 1.3–1.4 | 4.2–4.8 | [75] |
| Bovine, swine | 5 mL | ZON, α-ZOL, β-ZOL, ZAN, α-ZAL, β-ZAL | IAC | HPLC-ESI-MS/MS | 0.56–0.68 (CCa) | - | [90] |
| Bovine | 5 mL | ZON, α-ZOL, β-ZOL, ZAN | LLE: TBME, Hx, SPE: C18, SPE: -NH2, derivatization | GC-MS | CCa: 0.06–0.35 | CCb: 0.11–0.60 | [78] |
| Human | 5 mL | CIT, HO-CIT | IAC | LC-MS/MS | 0.02–0.05 | 0.05–0.1 | [87,88] |
| Human | 5 mL | DON, DOM-1 | IAC | LC-MS/MS | 0.10–0.16 | 0.2–0.3 | [85,86] |
| Human | 5 mL | OTA, OTA-GlcA, OTA-sulfates | LLE: CHCl3-Isopropanol | LC-MS/MS | 0.1–0.5 | 0.5–1 | [64] |
| Species | Volume (mL) | Mycotoxins | Extraction Procedure | Detection Technique | LOD (μg/L) | LOQ (μg/L) | Reference |
|---|---|---|---|---|---|---|---|
| Human | 10 | AFM1, FB1, FB2, OTA, OTα | IAC | HPLC-ESI-MS/MS | 0.001–0.045 | 0.004–0.135 | [82] |
| Human | 10 | AFB1, AFB2, AFG1, AFG2, OTA, DON, ZON, FB1, FB2, T-2, HT-2 | IAC | LC-QTRAP-MS/MS | 0.4–10 | 1.2–35 | [83] |
| Human Pig | 6 | AFM1, OTA, DON, DOM-1, α-ZOL, β-ZOL, FB1 | SPE: Oasis HLB, IAC | HPLC-Qtrap-MS/MS | 0.001–2.2 | 0.002–4.4 | [80] |
| Human Pig | 5 | DON, NEO, AFB1, AFM1, HT-2, HT2, OTA, OTα, ZON, α-ZOL, β-ZOL, FB1 | SALLE: MgSO4, EtOAc-ACN | LC-MS/MS | 0.01–0.5 | 0.07–3.3 | [30] |
| Pig | 6 | DON, DOM-1, OTA, AFB1, AFM1, FB1, ZON and α-ZOL | Myco6in1 IAC-Oasis HLB SPE | LC-MS/MS | - | - | [81] |
| Human | 10 | DON, OTA, FB1, AFB1, ZON, T-2, HT-2, AFB1, CIT, DOM, DON-2-GlcA, ZON-14-GlcA, α-ZOL, β-ZOL, 4-OH-OTA, OTα, AFM1, AFB1-N7-Gua | LLE: EtOaC/FA, SPE: SAX | LC-MS/MS | 0.01–3.65 | 0.02–5.76 | [79] |
| Human | 0.1 | DON, DON-3-GlcA, DON-15-GlcA, DOM-1, NIV, T-2, HT-2, ZON, ZON-14-O-GlcA, α-ZOL, b-ZOL, FB1, FB2, OTA, AFM1 | Dilute-shoot: ACN/H2O | HPLC-ESI-MS/MS | 0.005–2 | 0.017–6.7 | [71] |
| Human | - | DON, DON-3-GlcA, DON-15-GlcA, ZEN, ZEN-14-GlcA. | Dilute-shoot: ACN/H2O | LC-MS/MS | 0.2–4 | 0.3–6 | [104] |
| Human | 6 | DON, DOM-1, AFM1, FB1, ZON, α-ZOL, β-ZOL, OTA | SPE: Myco6in1® and OASIS® HLB columns | UPLC-MS/MS LC-QTrap MS/MS UPLC-API 5000 MS/MS | - | 0.02–4.4 0.006–9.9 | [77] |
| Human | 0.1 | AFM1, OTA, FB1, DON, DON-GlcAs, FB2, DOM-1, ZON, ZON-14-GlcA, α-ZOL, β-ZOL, T-2, HT-2, NIV | Dilute-shoot: ACN/H2O | LC-MS/MS | 0.05–12 | 0.15–40 | [70] |
| Human | 10 | LLE, SPE: AFB1, AFB2, AFG1, AFG2, AFB1-N7-gua, AFM1, CIT, DON, DON-3-GlcA, DOM-1, FB1, HFB1, OTA, OTα, 4-OH-OTA, T-2, HT-2, ZON, ZON-14-GlcA, α-ZOL, β-ZOL. Filter-shoot: AFB1, AFB2, AFG1, AFG2, AFM1, CIT, OH-CIT, DON, DON-3-GlcA , DON-15-GlcA, DOM-1, DOM-1-3-GlcA, 3-ADON, 3-ADON-15-GlcA, 15-ADON, 15-ADON-3-GlcA, DAS, FB1, FB2, FB3, FUS-X, OTA, OTα, T-2, HT-2, ZON, ZON-14-GlcA, α-ZOL, α-ZOL-7-GlcA, α-ZOL-14-GlcA, β-ZOL, β-ZOL-14-GlcA. | LLE: EtOAc/FA, SPE Filter-shoot | LC-MS/MS | - | - | [73,74] |
| Human | 10 | AFB1, DAS, FusX, 3-AcDON, 15-AcDON, β-ZEL, α-ZEL, CIT, OTα, DOM-1, FB1, FB2, FB3, DON, ZEN, T2, HT2, DON-3-GlcA, DOM-GlcA, ZEN-14-GlcA, β-ZEL-7-GlcA, β-ZEL-14- GlcA, α-ZEL-7-GlcA, α-ZEL-14-GlcA, 15-AcDON-3-GlcA, 3-AcDON-15-GlcA, OTA, CIT and AFM1 | Filter-shot IAC (OTA, CIT, AFM1) | LC-MS/MS | 0.001–0.2 | 0.003–0.5 | [105] |
| Human | 0.1 | DON, DON-3-GlcA, T-2, HT-2, HT-2-4-GlcA, FB1, AFB1, AFB2, AFG1, AFG2, AFM1, ZON, ZAN, α-ZOL, β-ZOL, ZON-14-GlcA, ZAN-14-GlcA, α-ZOL-14-GlcA, β-ZOL-14-GlcA, OTA, OTα, EN B, DH-CIT | Dilute-shoot: H2O/ACN/FA | LC-MS/MS | 0.0005–0.3125 | 0.0013–0.3125 | [68] |
| Human | 0.1 | DON, DON-3-GlcA, T-2, HT-2, HT-2-4-GlcA, FB1, AFB1, AFB2, AFG1, AFG2, AFM1, ZON, ZAN, α-ZOL, β-ZOL, ZON-14-GlcA, ZAN-14-GlcA, α-ZOL-14-GlcA, β-ZOL-14-GlcA, OTA, OTα, EN B, DH-CIT | Dilute-shoot: H2O/ACN/FA | LC-MS/MS | 0.000125–0.45 | 0.0005–0.9 | [69] |
| Human | 10 | DOM-1, DON, 3-ADON, FUS-X, DAS, NIV, NEO, HT-2, T-2, ZON, α-ZOL, β-ZOL, ZAN, α-ZAL, β-ZAL | QuECHERS, d-SPE | GC-MS/MS | 0.12–4 | 0.25–8 | [31,92,93] |
| Human | 1 | DON, DOM-1, 3-ADON, 15-ADON, ZON, α-ZOL, β-ZOL, ZAN, α-ZAL, β-ZAL | SALLE: ACN, NaCl-C18 | GC-MS/MS | 0.12–4 | 0.25–8 | [94] |
| Species | Sample | Volume | Mycotoxins | Extraction Procedure | Detection Technique | LOD (μg/L) | LOQ (μg/L) | Reference |
|---|---|---|---|---|---|---|---|---|
| Human | Breast milk | 1 mL | OTA | LLE: CHCl3 | HPLC-FLD | 0.01 | - | [106] |
| Human | Breast milk | 1 mL | AFM1, OTA | LLE: CHCl3, ACN | ELISA, HPLC-FD | - | - | [107] |
| Human | Breast milk | 5 mL | ZON | IAC | ELISA HPLC-FLD | 0.06 0.02–0.05 | - | [111] |
| Human | Breast milk | 10 mL | DON, 3-ADON, NIV, FUSX, NEO, DAS, HT-2, T-2, ZON, α-ZOL, β-ZOL, FB1, FB2, FB3, EN A, EN A1, EN B, EN B1, BEA, AFB1, AFB2, AFG1, AFG2, AFM1, STG, OTA, OTα | QuEChERS | UHPLC-HRMS | - | 1–50 | [110] |
| Cow | Milk | [24] | ||||||
| Donkey | Milk | AFM1 and AFM2 | IAC | HPLC-FLD, LC-MS/MS | [23] | |||
| Cow | Milk | 50 mL | AFM1 | IAC | LC-MS/MS | - | 0.01 | [22] |
| Human | Serum Urine | 1 mL 5mL | OTA, OTα | LLE: CHCl3/isopropanol | HPLC-FLD HPLC-ESI-MS/MS | 0.07 0.02 | 0.1 0.5 | [95,96] |
| Human | Serum Urine | 1 mL 5 mL | CIT | LLE: ACN, IAC: CitriTest | HPLC-FLD | [96,97,98,99] | ||
| Human | Serum Urine | 250 μL 5 mL | ENs, BEA | LLE: MeOH/H2O, SPE: GCB | LC-MS/MS | 0.01–0.0025–0.02 | 0.02–0.04 0.005–0.02 | [98] |
| Chicken Pig | Serum Bile | 250 μL 1 mL | DON, DOM-1, T-2, HT-2 | LLE: MeOH, SPE LLE: MeOH/H2O, EtOAc | LC-MS/MS | 0.01–0.63 | 1.0–2.5 | [112,113] |
| Pig | Serum, urine, liquor | 500 μL | ZON, DON, ZAN, α-ZOL, β-ZOL,α-ZAL, β-ZAL | SPE: Oasis HLB | HPLC-ESI-MS/MS | 0.005–0.71 0.03–0.16 0.02–0.21 | 0.08–2.37 0.1–0.52 0.07–0.70 | [97] |
| Rat | Serum, urine, feces | 200 μL 100 mg | EN A | LLE: EtOAc | LC-MS/MS | 1.8–2.3 | 5.4–7 | [101] |
| Rat | Serum, urine, feces | 500 μL 500 mg | EN A, A1, B, B1 | LLE: ACN | LC-MS/MS | 0.2–1 | 2–10 | [100] |
| Horse | Serum, urine, feces | 1 mL 5 mL 2 g | ZON, DON, ZAN, α-ZOL, β-ZOL, α-ZAL, β-ZAL | IAC SPE: C18 , IAC SPE: C18 , IAC | HPLC-APCI-MS/MS | 0.1–0.3 0.1–0.2 0.1–0.5 | 0.5–0.6 0.5–1 0.5–1 | [102] |
| Human | Feces | 1–2 g 1 mL | OTA, OTB | SPE: C18 | HPLC-FLD | - | 1.25–2.22 1.44–2.99 | [108] |
| Human | Breast milk | 2 mL | AFB1, AFB2, AFG1, AFG2, AFM1, OTA | LLE: ACN/EtOAc, LTP (low temperature purification) | HPLC-FLD LC-MS/MS | - | 0.005–0.03 | [109] |
| Species | Biological Sample | Sample Size | Mycotoxins | Extraction Procedure | Detection Technique | LOD (μg/kg) | LOQ (μg/kg) | Reference |
|---|---|---|---|---|---|---|---|---|
| Mouse | Plasma, spleen, liver, lung, and kidney | 40–200 mg | DON | PBS | ELISA | - | - | [19] |
| Mouse | Plasma, liver, kidney, heart, spleen, and brain | 100 μL (extract) | DON | ice-cold ethanol/trichloroacetic acid | ELISA | - | - | [139] |
| Rat | Plasma, liver, kidney | 250 μL 200–400 mg | OTA | LLE: ACN, SPE | HPLC-FLD | 1–14.3 | 8.4–52.8 | [121] |
| Pig | Plasma, liver, kidney | 800 μL 20 g | OTA | LLE: ACN, SPE | HPLC-FD and LC-MS/MS | 0.14 0.25 | 0.25 0.5 | [120] |
| Swine, cattle, sheep, horse, fish, chicken, turkey, geese, duck | Muscles, liver and kidneys | 10 g | OTA | LLE: CHCl3, IAC: OchraTest | LC-FD | - | 0.2 | [134] |
| Laying Hens | Kidneys, liver, bile (eggs) | 2.5 g 200 μL | OTA | LLE: CHCl3, SPE | HPLC-FD | 0.3–0.5 | 1 | [27] |
| Turkey poults | Plasma, muscle, liver, and kidney | 250 μL 1 g | FB1 | SPE: SAX IAC: FumoniPrep | HPLC-FLD | 13 | 25 | [138] |
| Rat | Serum, bile, and urine. Lung, liver, spleen, kidneys, heart, testes, brain, muscle, adipose tissue, stomach, and small intestine | ZAN | LLE, IAC | LC-MS/MS HPLC-FD | - | 0.5 10 | [122] | |
| Rat | Liver and kidney | 1 g | T-2 | LLE: ACN, SPE: charcoal/celite/aluminium trioxide | HPTLC | - | 100 | [123] |
| Rat | Serum, stomach, duodenum, jejunum, ileum, colon, liver | 0.5 g 0.5 mL | EN A | LLE: EtOAc | LC-MS/MS | 200 | 600 | [118] |
| Chicken | Serum, liver, kidney, heart, muscle, small intestine, and excreta | 1 mL 5 g | NIV | LLE: ACN/H2O (NH4)2SO4, SPE C18 | LC-MS/MS | - | 2–2.5 | [125] |
| Cow | Serum, milk, liver, kidney, muscles, fat, and jejuno, ileum | 2–5 mL 10 g | OTA | LLE: ACN/Hex, IAC | HPLC-FD | - | - | [133] |
| Species | Biological Sample | Sample Size | Mycotoxins | Extraction Procedure | Detection Technique | LOD (μg/kg) | LOQ (μg/kg) | Reference |
|---|---|---|---|---|---|---|---|---|
| Swine | Plasma, liver | 10 g | FB1, AP-1 | LLE: ACN, SPE: C18, SAX, Oasis HLB. LLE: ACN/MeOH, Hx, IAC: FumoniPrep | HPLC-FD | 10–20 | 42–75 | [132] |
| Pig | Plasma, bile, urine, liver, kidney, muscle | 1.5 mL 1 mL 1 mL 2 g 2.4 g | DON, DOM-1 | LLE:Cl3, IAC: DON-test | LC-MS/MS | 1.5–10 | 2–10 | [137] |
| Goat | Plasma, urine, feces, liver | 5 mL 5 g | ZAN and metabolites | LLE: EtOAc, IAC: Easi-Extract ZAN | HPLC | - | 2.1–46.6 | [135] |
| Bovine | Mucle, liver | 5 g | ZON, α-ZOL, β-ZOL, ZAN, α-ZAL, β-ZAL | LLE: MeOH-Hx, SPE: Sep-pak amino | UPLC-MS/MS; CLEIA | 0.5 | 0.5–0.7 | [131] |
| Boar | Muscle, liver, kidney, spleen, cardiac muscle, lung, ovary, uterus | 3 g | ZON, α-ZOL, β-ZOL | LLE: MeOH, IAC | LC-MS | 1 | - | [136] |
| Pig | Plasma, fat, muscle, stomach, brain, small intestines, heart, lung, spleen, urine, feces | 0.5 mL 2 g | T-2, HT-2, T-2 triol | LLE: ACN. LLE: EtOAc, SPE: Varian Bond-Elut | LC-MS/MS | 0.3–2 | 1–5 | [126] |
| Broiler, poultry | Liver and meat | 5 g | ENs, BEA | LLE, SPE | LC-MS/MS | 0.015–0.56 | 0.03–1.12 | [125] |
| Fish | Liver, viscera, tissue, head | 5–10 g | ENs | LLE: ACN, SPE: C18 | LC-MS/MS | 0.3–3 | 1–10 | [128] |
| Mice | Liver, kidney, colon, fat, brain, muscle, tumor urine, serum | 0.2 g 50 μL | BEA, EN B | LLE: ACN | LC-MS/MS | - | 0.05–0.15 | [26] |
| Mice | Serum, Brain | 50 μL | BEA, ENs | LLE: ACN-H2O | UPLC-MS/MS | 0.3 | - | [115] |
| Species | Biological Sample | Sample Size | Mycotoxins | Extraction Procedure | Detection Technique | LOD (μg/kg) | LOQ (μg/kg) | Reference |
|---|---|---|---|---|---|---|---|---|
| Human | Urine and nasal secretions (nasal washes, sputa), heart, liver, urine | TCT, AFs, OTA | PBS, formalin | ELISA, Fluorometry | 0.2–2 | - | [114] | |
| Human Rat Dogs Rabbit | Human: urine, blood, feces, saliva, nasal secretions, breast milk, amniotic fluid of pregnant women. Animal: liver, spleen, lung, kidney, stomach, colon, brain, urine, blood, and feces. | 200 μL 200 μg | AFB1, AFB2, AFG1, AF2, AFM1, AFM2, OTA, DON, NIV, T-2, HT-2, 3-ADON, 15-ADON, NEO, FUS-X, DAS, MAS, ZON, ZAN, α-ZOL, β-ZOL, α-ZAL, β-ZAL, T-2 triol, T-2 tertraol, DOM-1, FB1, FB2 | PLE: ACN/H2O/hx/acetic acid | HPLC-MS/MS | CCa: 0.01–0.69 | 0.2–0.5 CCb: 0.15–1.26 | [28] |
| Chicken Pig | Muscle, liver | 1g | T-2, HT-2, T-2-triol, NEO, DON, 3-Ac-DON, 5-Ac-DON, DOM, NIV | LLE: ACN/EtOAc SPE: Oasis HLB | UPLC-MS/MS | 3–15 | 10–50 | [129] |
| Rat | Plasma, liver, kidney | 100 μL (25 mg tissue) | ABF1, OTA | LLE: CHCl3, IAC | UHPLC-FLD | 0.01–0.3 | 2–8 | [117] |
| Rat | Plasma, urine, liver, kidney, bladder, spleen, lung, stomach, small intestine, large intestine | - | DON-Glc, DON-GlcAs, ZON-14-Glc, ZON-14-GlcA, 3-ADON, 15-ADON | LLE: ACN | UPLC-MS/MS | 0.3–16.3 | 0.6–54.4 | [116] |
| Chicken | Muscle, liver, kidney, fat, tissues | 2 g | DON, 3-ADON, 15-ADON, DOM-1 | LLE: EtOAc, SPE: Oasis HLB | LC–MS/MS | CCa: 0.16–0.92 | CCb: 0.68–2.07 | [130] |
| Chickens | Muscle and liver | 1 g | NIV, DON, DOM, NEO, 3-ADON, 15-ADON, T-2-triol, HT-2, T-2 | LLE: ACN/H2O, SPE: Oasis HLB, IAC: charcoal/alumina/celite | UPLC-MS/MS | 1–5 | 3–15 | [129] |
| Broiler | Heart, liver, spleen, lung, kidney, Glandular stomach, muscular stomach, small intestine, muscle, bone, brain | 1 g | T-2, HT-2, DAS | LLE: EtOAc | LC-MS/MS | 0.02–0.05 | 0.08–0.17 | [119] |
| Pig | Plasma, urine feces, liver, kidney, spleen, muscle, intestine, bile | 1 mL 5 g | FUS-X, NIV | LLE: ACN/H2O, SPE: C18 | LC-MS/MS | 1.0–1.8 | 1.11–2.4 | [124] |
| Pig | Bile, liver, and muscle | - | ZON, α-ZOL, β-ZOL, DON | LLE: MeOH/H2O, Hx, SPE: Oasis HLB | HPLC and EIA | - | - | [18] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escrivá, L.; Font, G.; Manyes, L.; Berrada, H. Studies on the Presence of Mycotoxins in Biological Samples: An Overview. Toxins 2017, 9, 251. https://doi.org/10.3390/toxins9080251
Escrivá L, Font G, Manyes L, Berrada H. Studies on the Presence of Mycotoxins in Biological Samples: An Overview. Toxins. 2017; 9(8):251. https://doi.org/10.3390/toxins9080251
Chicago/Turabian StyleEscrivá, Laura, Guillermina Font, Lara Manyes, and Houda Berrada. 2017. "Studies on the Presence of Mycotoxins in Biological Samples: An Overview" Toxins 9, no. 8: 251. https://doi.org/10.3390/toxins9080251