Synthetic α-Conotoxin Mutants as Probes for Studying Nicotinic Acetylcholine Receptors and in the Development of Novel Drug Leads

Abstract
:Abbreviations
| Ac: | Aplysia californica |
| AChBP: | acetylcholine binding protein |
| Bt: | Bulinus truncatus |
| GABA: | γ-aminobutyric acid |
| HEPES: | (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) |
| Laa: | lipidic amino acid |
| Ls: | Lymnaea stagnalis |
| nAChR: | nicotinic acetylcholine receptor |
| NET: | norepinephrine transporter |
| PS-SCL: | positional scan synthetic combinatorial library |
| SCAL: | safety catch amide linker |
| Sec: | selenocysteine |
| SPPS: | solid-phase peptide synthesis |
1. Introduction
2. α-Conotoxins as Probes for Nicotinic Acetylcholine Receptors

3. Structural Studies of α-Conotoxins


{kind=link}
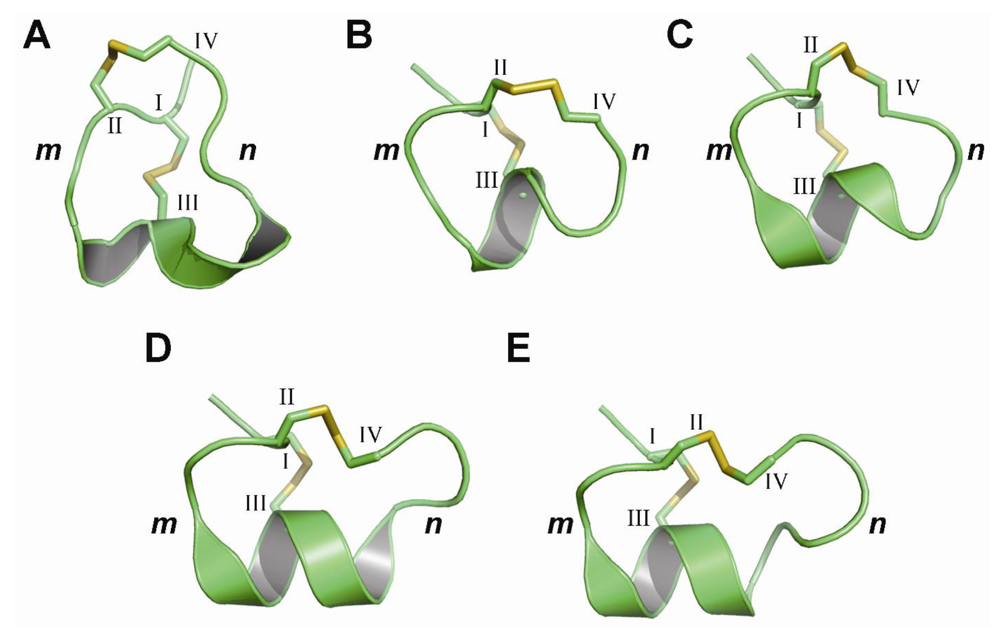{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |

4. Synthetic Mutants of α-Conotoxins
4.1. α-Conotoxins ImI and ImII


4.2. α-Conotoxins PnIA, PnIA and TxIA
4.3. α-Conotoxin MII
4.4. α-Conotoxin GID
4.5. α-Conotoxin ArIB
4.6. α-Conotoxins RgIA and Vc1.1
5. High Throughput Synthesis of α-Conotoxin Analogs
6. Novel α-Conotoxin Analogs with Enhanced Pharmacokinetic Properties

7. Conclusions and Future Perspectives
References
- Endean, R.; Rudkin, C. Studies of the venoms of some Conidea. Toxicon 1963, 1, 49–64. [Google Scholar]
- McIntosh, J.M.; Jones, R.M. Cone venom-from accidental stings to deliberate injection. Toxicon 2001, 39, 1477–1451. [Google Scholar]
- Olivera, B.M.; Rivier, J.; Clark, C.; Corpuz, G.P.; Mena, E.E.; Ramilo, C.A.; Cruz, L.J. Diversity of Conus neuropeptides. Science 1990, 249, 257–263. [Google Scholar]
- Cruz, L.J.; Gray, W.R.; Olivera, B.M. Purification and properties of a myotoxin from Conus geographus venom. Arch. Biochem. Biophys. 1978, 190, 539–548. [Google Scholar]
- Gray, W.R.; Luque, A.; Olivera, B.M.; Barret, J.; Cruz, L.J. Peptide toxins from Conus geographus venom. J. Biol. Chem. 1981, 256, 4734–4740. [Google Scholar]
- Sollod, B.; Wilson, D.; Zhaxybayeva, O.; Gogarten, J.P.; Drinkwater, R.; King, G.F. Were arachnids the first to use combinatorial peptide libraries. Peptides 2005, 26, 131–139. [Google Scholar]
- Olivera, B.M. Conus peptides: Biodiversity-based discovery and exogenomics. J. Biol. Chem. 2006, 281, 31173–31177. [Google Scholar]
- Han, T.S.; Teichert, R.W.; Olivera, B.M.; Bulaj, G. Conus venoms - A rich source of peptide-based therapeutics. Curr. Pharm. Design 2008, 14, 2462–2479. [Google Scholar]
- Armishaw, C.J.; Alewood, P.F. Conotoxins as research tools and drug leads. Curr. Protein Pept. Sci. 2005, 6, 221–240. [Google Scholar]
- Miljanich, G.P. Ziconotide: Neuronal calcium channel blocker for treating severe chronic pain. Curr. Med. Chem. 2004, 11, 3029–3040. [Google Scholar]
- Nicke, A.; Wonnacott, S.; Lewis, R.J. α-Conotoxins as tools for the elucidation of structure and function of neuronal nicotinic acetylcholine receptor subtypes. Eur. J. Biochem. 2004, 271, 2305–2319. [Google Scholar]
- Sine, S.M.; Engel, A.G. Recent advances in Cys-loop receptor structure and function. Nature 2006, 440, 455–463. [Google Scholar]
- Romanelli, M.N.; Gratteri, P.; Guandalini, L.; Martini, E.; Bonaccini, C.; Gualtieri, F. Central Nicotinic Receptors: Structure, Function, Ligands, and Therapeutic Potential. Chem. Med. Chem. 2007, 2, 746–767. [Google Scholar]
- Jensen, A.; Frølund, B.; Liljefors, T.; Krogsgaard-Larsen, P. Neuronal nicotinic acetylcholine receptors: Structural revelations, target identifications and therapeutic inspirations. J. Med. Chem. 2005, 48, 4705–4744. [Google Scholar] [CrossRef] [PubMed]
- Sher, E.; Chen, Y.; Sharples, T.J.; Broad, L.M.; Benedetti, G.; Zwart, R.; McPhie, G.I.; Pearson, K.H.; Baldwinson, T.; DeFillipi, G. Physiological Roles of Neuronal Nicotinic Receptors Subtypes: New Insights on the Nicotinic Modulation of Neurotransmitter Release, Synaptic Transmission and Plasticity. Curr. Top. Med. Chem. 2004, 4, 283–297. [Google Scholar]
- Changeux, J.P.; Edelstein, S.J. Allosteric mechanisms of signal transduction. Science 2005, 308, 1424–1428. [Google Scholar]
- Taly, A.; Corringer, P.J.; Guedin, D.; Lestage, P.; Changeux, J.P. Nicotine receptors: Allosteric transitions and therapuetic targets in the nervous system. Nat. Rev. Drug Discov. 2009, 8, 733–750. [Google Scholar]
- Lukas, R.J.; Changeux, J.P.; Le Novere, N.; Albuquerque, E.X.; Balfour, D.J.K.; Berg, D.K.; Bertrand, D.; Chiappinelli, V.A.; Clarke, P.B.S.; Collins, A.C.; Dani, J.A.; Grady, S.R.; Kellar, K.J.; Lindstrom, J.M.; Marks, M.J.; Quik, M.; Taylor, P.W.; Wonnacott, S. International union of pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol. Rev. 1999, 51, 397–401. [Google Scholar] [PubMed]
- Arneric, S.P.; Holladay, M.W.; Williams, M. Neuronal nicotinic receptors: A perspective on two decades of drug discovery research. Biochem. Pharmacol. 2007, 74, 1092–1101. [Google Scholar]
- Niaura, R.; Jones, C.; Kirkpatrick, P. Varenicline. Nat. Rev. Drug Discov. 2006, 5, 537–538. [Google Scholar]
- Mihalak, K.B.; Carroll, F.I.; Luetje, C.W. Varenicline is a partial agonist at α4β2 and a full agonist at α7 neuronal nicotinic receptors. Mol. Pharmacol. 2006, 70, 801–805. [Google Scholar]
- Moore, T.J.; Furberg, C.D. Risk of psychiatric side effects with varenicline. Brit. Med. J. 2009, 339, b4964. [Google Scholar]
- Kuehn, B.M. Studies linking smoking-cessation drug with suicide risk spark concerns. J. Am. Med. Assoc. 2009, 301, 1007–1008. [Google Scholar]
- Livett, B.G.; Sandall, D.W.; Keays, D.; Down, J.; Gayler, K.R.; Satkunanathan, N.; Khalil, Z. Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon 2006, 48, 810–829. [Google Scholar]
- Martinez, J.S.; Olivera, B.M.; Gray, W.R.; Craig, A.G.; Groebe, D.R.; Abramson, S.N.; McIntosh, J.M. α-Conotoxin EI, a new nicotinic acetylcholine receptor antagonist with novel selectivity. Biochemistry 1995, 34, 14519–14526. [Google Scholar]
- Nicke, A.; Loughnan, M.L.; Millard, E.L.; Alewood, P.F.; Adams, D.J.; Daly, N.L.; Craik, D.J.; Lewis, R.J. Isolation, Structure, and Activity of GID, a Novel α4/7-Conotoxin with an Extended N-terminal Sequence. J. Biol. Chem. 2003, 278, 3137–3144. [Google Scholar]
- Whiteaker, P.; Christensen, S.; Yoshikami, D.; Dowell, C.; Watkins, M.; Gulyas, J.; Rivier, J.; Olivera, B.M.; McIntosh, J.M. Discovery, synthesis, and structure activity of a highly selective alpha7 nicotinic acetylcholine receptor antagonist. Biochemistry 2007, 46, 6628–6638. [Google Scholar]
- Lopez-Vera, E.; Aguilar, M.B.; Schiavon, E.; Marinzi, C.; Oritz, E.; Restano Cassulini, R.; Batista, C.V.F.; Possani, L.D.; Heimer de lat Cotera, E.P.; Peri, F.; Becerril, B.; Wanke, E. Novel α-conotoxins from Conus spurius and the α-conotoxin EI share high-affinity potentiation and low-affinity inhibition of nicotinic acetylcholine receptors. FEBS J. 2007, 274, 3972–3985. [Google Scholar]
- Dowell, C.; Olivera, B.M.; Garret, J.E.; Staheli, S.T.; Watkins, M.; Kuryatov, A.; Yoshikami, D.; Lindstrom, J.M.; McIntosh, J.M. α-Conotoxin PIA is selective for α6 subunit-containing nicotinic acetylcholine receptors. J. Neurosci. 2003, 23, 8445–8452. [Google Scholar]
- McIntosh, J.M.; Santos, A.D.; Olivera, B.M. Conus Peptides Targeted to Specific Nicotinic Acetylcholine Receptor Subtypes. Annu. Rev. Biochem. 1999, 68, 59–88. [Google Scholar]
- Loughnan, M.; Alewood, P.F. Physico-chemical characterization and synthesis of neuronally active a-conotoxins. Eur. J. Biochem. 2004, 271, 2294–2304. [Google Scholar]
- Norton, R.S.; Olivera, B.M. Conotoxins down under. Toxicon 2006, 48, 780–798. [Google Scholar]
- Peng, C.; Han, Y.; Sanders, T.; Chew, G.; Liu, J.; Hawrot, E.; Chi, C.; Wang, C. α4/7-conotoxin Lp1.1 is a novel antagonist of neuronal nicotinic acetylcholine receptors. Peptides 2008, 29, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Akondi, K.B.; Zhangsun, D.; Wu, Y.; Zhu, X.; Hu, Y.; Christensen, S.; Dowell, C.; Daly, N.; Craik, D.J.; Wang, C.-I.; Lewis, R.J.; Alewood, P.F.; McIntosh, J.M. The atypical α-conotoxin LtIA from Conus litteratus targets a novel microsite of the alpha3beta2 nicotinic receptor. J. Biol. Chem. 2010, 285, 12355–12366. [Google Scholar]
- Marshall, I.G.; Harvey, A.L. Selective neuromuscular blocking properties of α-conotoxins In vivo. Toxicon 1990, 28, 231–234. [Google Scholar]
- Myers, R.A.; Cruz, L.J.; Rivier, J.E.; Olivera, B.M. Conus peptides as chemical probes for receptors and ion channels. Chem. Rev. 1993, 93, 1923–1936. [Google Scholar]
- Lui, L.; Chew, G.; Hawrot, E.; Chi, C.; Wang, C. Two potent α3/5 conotoxins from piscivorous Conus achatinus. Acta Biochim. Biophys. Sinica. 2007, 39, 438–444. [Google Scholar]
- Lopez-Vera, E.; Jacobsen, R.B.; Ellison, M.; Olivera, B.M.; Teichert, R.W. A novel alpha-conotoxin (alpha-PIB) isolated from C. purpurascens is selective for skeletal muscle nicotinic acetylcholine receptors. Toxicon 2007, 49, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.M.; Cruz, L.J.; Hunkapiller, M.W.; Gray, W.R.; Olivera, B.M. Isolation and structure of a peptide toxin from the marine snail Conus magus. Arch. Biochem. Biophys. 1982, 218, 329–334. [Google Scholar]
- Zafaralla, G.C.; Ramilo, C.; Gray, W.R.; Karlstrom, R.; Olivera, B.M.; Cruz, L.J. Phylogenetic specificity of cholinergic ligands: α-Conotoxin SI. Biochemistry 1988, 27, 7102–7105. [Google Scholar]
- Myers, R.A.; Zafarella, G.C.; Gray, W.R.; Abbot, J.; Cruz, L.J.; Olivera, B.M. α-Conotoxins, small peptide probes of nicotinic acetylcholine receptors. Biochemistry 1991, 30, 9370–9377. [Google Scholar]
- Ramilo, C.; Zafaralla, G.C.; Nadasdi, L.; Hammerland, L.G.; Yoshikami, D.; Gray, W.R.; Kristipati, R.; Ramachandran, J.; Miljanich, G.; Olivera, B.M.; Cruz, L.J. Novel α- and ω-conotoxins from Conus striatus venom. Biochemistry 1992, 31, 9919–9926. [Google Scholar]
- Favreau, P.; Krimm, I.; Le Gall, F.; Bobenreith, M.; Lamthanh, H.; Bouet, F.; Servent, D.; Molgo, J.; Menez, A.; Letourneux, Y.; Lancelin, J. Biochemical characterization and nuclear magnetic resonance structure of novel α-conotoxins isolated from the venom of Conus consors. Biochemistry 1999, 38, 6317–6326. [Google Scholar]
- Luo, S.; Kulak, J.M.; Cartier, G.E.; Jacobsen, R.B.; Yoshikami, D.; Olivera, B.M.; McIntosh, J.M. α-Conotoxin AuIB selectively blocks α3β4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J. Neurosci. 1998, 18, 8571–8579. [Google Scholar]
- Azam, L.; Dowell, C.; Watkins, M.; Stitzel, J.A.; Olivera, B.M.; McIntosh, J.M. α-Conotoxin BuIA, a novel peptide from Conus bullatus distinguishes among neuronal nicotinic acetylcholine receptors. J. Biol. Chem. 2005, 280, 80–87. [Google Scholar]
- McIntosh, J.M.; Yoshikami, D.; Mahe, E.; Nielsen, D.B.; Rivier, J.E.; Gray, W.R.; Olivera, B.M. A nicotinic acetylcholine receptor ligand of unique specificity, α-conotoxin ImI. J. Biol. Chem. 1994, 269, 16733–16739. [Google Scholar]
- Ellison, M.; McIntosh, J.M.; Olivera, B.M. α-Conotoxins ImI and ImII. J. Biol. Chem. 2003, 278, 757–764. [Google Scholar]
- Ellison, M.; Haberlandt, C.; Gomez-Casati, M.E.; Watkins, M.; Elgoyhen, A.B.; McIntosh, J.M.; Olivera, B.M. α-RgIA: A novel conotoxin that specifically and potently blocks the α9α10 nAChR. Biochemistry 2006, 45, 1511–1517. [Google Scholar]
- Santos, A.D.; McIntosh, J.M.; Hillyard, D.R.; Cruz, L.J.; Olivera, B.M. The A-superfamily of conotoxins. J. Biol. Chem. 2004, 279, 17596–17606. [Google Scholar]
- Yuan, D.D.; Han, Y.H.; Wang, C.G.; Chi, C.W. From the identification of gene organization of alpha conotoxins to the cloning of novel toxins. Toxicon 2007, 49, 1135–1149. [Google Scholar]
- Jin, A.-H.; Daly, N.L.; Nevin, S.T.; Wang, C.I.A.; Dutertre, S.; Lewis, R.J.; Adams, D.J.; Craik, D.J.; Alewood, P.F. Molecular engineering of conotoxins: The importance of loop size to α-conotoxin structure and function. J. Med. Chem. 2008, 51, 5575–5584. [Google Scholar]
- Janes, R.W. α-Conotoxins as selective probes for nicotinic acetylcholine receptor subclasses. Curr. Opin. Pharmacol. 2005, 5, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, J.A.; Keays, D.A.; Kelley, W.P.; Sandall, D.W.; Bingham, J.-P.; Livett, B.G.; Gayler, K.R.; Sweedler, J.V. Determining sequences and posttranslational modifications of novel conotoxins from Conus victoriae using cDNA sequencing and mass spectrometry. J. Mass. Spec. 2004, 39, 548–557. [Google Scholar]
- Franco, A.; Pisarewicz, K.; Moller, C.; Mora, D.; Fields, G.B.; Mari, F. Hyperhydroxylation: a new strategy for neuronal targeting by venomous marine molluscs. Prog. Mol. Subcell. Biol. 2006, 43, 83–103. [Google Scholar]
- Loughnan, M.; Bond, T.; Atkins, A.; Cuevas, J.; Adams, D.J.; Broxton, N.M.; Livett, B.G.; Down, J.G.; Jones, A.; Alewood, P.F.; Lewis, R.J. α-Conotoxin EpI, a novel sulfated peptide from Conus episcopatus that selectively targets neuronal nicotinic acetylcholine receptors. J. Biol. Chem. 1998, 273, 15667–15674. [Google Scholar]
- Wolfender, J.L.; Chu, F.X.; Ball, H.; Wolfender, F.; Fainzilber, M.; Baldwin, M.A.; Burlingame, A.L. Identification of tyrosine sulfation in Conus pennaceus conotoxins a-PnIA and a-PnIB: Further investigation of labile sulfo- and phosphopeptides by electrospray, matrix-assisted laser desorption/ionization (MALDI) and atmospheric pressure MALDI mass spectrometry. J. Mass. Spec. 1999, 34, 447–454. [Google Scholar]
- Loughnan, M.L.; Nicke, A.; Jones, A.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Chemical and functional identification and charactrisation of novel sulfated α−conotoxins from the cone snail Conus anenome. J. Med. Chem. 2004, 47, 1234–1241. [Google Scholar]
- Craig, A.G.; Bandyopadhyay, P.; Olivera, B.M. Posttranslationally modified neuropeptides from Conus venoms. Eur. J. Biochem. 1999, 264, 271–275. [Google Scholar]
- Nicke, A.; Samochocki, M.; Loughnan, M.L.; Bansal, P.S.; Maelicke, A.; Lewis, R.J. α-Conotoxins EpI and AuIB switch subtype selectivity and activity in native versus recombinant nicotinic acetylcholine receptors. FEBS Lett. 2004, 554, 219–223. [Google Scholar]
- Millard, E.L.; Daly, N.L.; Craik, D.J. Structure-activity relationships of α-conotoxins targeting neuronal nicotinic acetylcholine receptors. Eur. J. Biochem. 2004, 271, 2320–2326. [Google Scholar]
- Guddat, L.W.; Martin, J.A.; Shan, L.; Edmundson, A.B.; Gray, W.R. Three-dimensional structure of the α-conotoxin GI at 1.2Å resolution. Biochemistry 1996, 35, 11329–11355. [Google Scholar] [PubMed]
- Hu, S.H.; Gehrmann, J.; Guddat, L.W.; Alewood, P.F.; Craik, D.J.; Martin, J.L. The 1.1Å crystal structure of the neuronal acetylcholine receptor antagonist, a-conotoxin PnIA from Conus pennaceus. Structure 1996, 4, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.-H.; Gehrmann, J.; Alewood, P.F.; Craik, D.J.; Martin, J.L. Crystal structure at 1.1Å resolution of α-conotoxin PnIB: Comparison with α-conotoxins PnIA and GI. Biochemistry 1997, 36, 11323–11330. [Google Scholar] [CrossRef] [PubMed]
- Marx, U.C.; Daly, N.L.; Craik, D.J. NMR of conotoxins: structural features and an analysis of chemical shifts of posttranslationally modified amino acids. Magn. Reson. Chem. 2006, 44, S41–S50. [Google Scholar]
- Hill, J.M.; Oomen, C.J.; Miranda, L.P.; Bingham, J.P.; Alewood, P.F.; Craik, D.J. Three-dimensional solution structure of α-sonotoxin MII by NMR spectroscopy: effects of solution environment on helicity. Biochemistry 1998, 37, 15621–15630. [Google Scholar]
- Maslannikov, I.V.; Shenkarev, Z.O.; Zhmak, M.N.; Ivanov, V.T.; Methfessel, C.; Tsetlin, V.I.; Arseniev, A.S. NMR spatial structure of α-conotoxin ImI reveals a common scaffold in snail and snake toxins recognizing neuronal nicotinic acetylcholine receptors. FEBS Lett. 1999, 444, 275–280. [Google Scholar]
- Jin, A.-H.; Brandstaetter, H.; Nevin, S.T.; Tan, C.C.; Clark, R.J.; Adams, D.J.; Alewood, P.F.; Craik, D.J.; Daly, N.L. Structure of α-conotoxin BuIA: influences of disulfide connectivity on structural dynamics. BMC Struct. Biol. 2007, 7, 28–41. [Google Scholar]
- Cho, J.-H.; Mok, K.H.; Olivera, B.M.; McIntosh, J.M.; Park, K.-H.; Han, K.-H. Nuclear magnetic resonance solution conformation of α-conotoxin AuIB, an α3β4 subtype-selective neuronal nicotinic acetylcholine receptor antagonist. J. Biol. Chem. 2000, 275, 8680–8685. [Google Scholar]
- McIntosh, J.M.; Dowell, C.; Watkins, M.; Garrett, J.E.; Yoshikami, D.; Olivera, B.M. α-conotoxin GIC from Conus geographus, a novel peptide antagonist of nicotinic acetylcholine receptors. J. Biol. Chem. 2002, 277, 33610–33615. [Google Scholar] [PubMed]
- Cartier, G.E.; Yoshikami, D.; Gray, W.R.; Luo, S.; Olivera, B.M.; McIntosh, J.M. A new α-conotoxin which targets α3β2 nicotinic acetylcholine receptors. J. Biol. Chem. 1996, 271, 7522–7528. [Google Scholar]
- Talley, T.T.; Olivera, B.M.; Han, K.-H.; Christensen, S.B.; Dowell, C.; Tsigelny, I.; Ho, K.-Y.; Taylor, P.; McIntosh, J.M. α-Conotoxin OmIA is a potent ligand for the acetylcholine-binding protein as well as a3b2 and a7 nicotinic acetylcholine receptors. J. Biol. Chem. 2006, 281, 24678–24686. [Google Scholar] [PubMed]
- McIntosh, J.M.; Plazas, P.V.; Watkins, M.; Gomez-Casati, M.E.; Olivera, B.M.; Elgoyhen, A.B. A Novel α-Conotoxin, PeIA, cloned from Conus pergrandis, discriminates between rat α9α10 and α7 nicotinic cholinergic receptors. J. Biol. Chem. 2005, 280, 30107–30112. [Google Scholar]
- Fainzilber, M.; Hasson, A.; Oren, R.; Burlingame, A.L.; Gordon, D.; Spira, M.E.; Zlotkin, E. New mollusc-specific α-conotoxins block Aplysia neuronal acetylcholine receptors. Biochemistry 1994, 33, 9523–9529. [Google Scholar]
- Dutertre, S.; Ulens, C.; Büttner, R.; Fish, A.; van Elk, R.; Kendel, Y.; Hopping, G.; Alewood, P.F.; Schroeder, C.; Nicke, A.; Smit, A.B.; Sixma, T.K.; Lewis, R.J. AChBP-targeted α-conotoxin correlates distinct binding orientations with nAChR subtype selectivity. EMBO J. 2007, 26, 3858–3867. [Google Scholar]
- Sandall, D.W.; Satkunanathan, N.; Keays, D.A.; Polidano, M.A.; Liping, X.; Pham, V.; Down, J.G.; Khalil, Z.; Livett, B.G.; Gayler, K.R. A novel α-Conotoxin identified by gene sequencing is active in supressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry 2003, 42, 6904–6911. [Google Scholar]
- Quiram, P.A.; Sine, S.M. Structural elements in α-conotoxin ImI essential for binding to neuronal α7 receptors. J. Biol. Chem. 1998, 273, 11007–11011. [Google Scholar]
- Jacobsen, R.B.; DelaCruz, R.G.; Gros, J.H.; McIntosh, J.M.; Yoshikami, D.; Olivera, B.M. Critical residues influence the affinity and selectivity of α-conotoxin MI for nicotinic acetylcholine receptors. Biochemistry 1999, 38, 13310–13315. [Google Scholar]
- Lamthanh, H.; Jegou-Matheron, C.; Servent, D.; Menez, A.; Lancelin, J. Minimal conformation of the alpha-conotoxin ImI for the α7 neuronal nicotinic acetylcholine receptor recognition: correlated CD, NMR and binding studies. FEBS Lett. 1999, 454, 293–298. [Google Scholar]
- Everhart, D.; Cartier, G.E.; Malhotra, A.; Gomes, A.V.; McIntosh, J.M.; Luetje, C.W. Determinants of potency on α-conotoxin MII, a peptide antagonist of neuronal nicotinic receptors. Biochemistry 2004, 43, 2732. [Google Scholar]
- Kang, T.S.; Radic, Z.; Talley, T.T.; Jois, S.D.S.; Taylor, P.; Kini, R.M. Protein folding determinants: Structural features determining alternative disulfide pairing in α- and χ/λ-Conotoxins. Biochemistry 2007, 46, 3338–3355. [Google Scholar]
- Gehrmann, J.; Alewood, P.F.; Craik, D.J. Structure determination of the three disulfide bond isomers of α-conotoxin GI: A model for the role of disulfide bonds in structural stability. J. Mol. Biol. 1998, 278, 401–415. [Google Scholar]
- Dutton, J.L.; Bansal, P.S.; Hogg, R.C.; Adams, D.J.; Alewood, P.F.; Craik, D.J. A new level of conotoxin diversity, a non-native disulfide bond connectivity in α-conotoxin AuIB reduces structural definition but increases biological activity. J. Biol. Chem. 2002, 277, 48849–48857. [Google Scholar]
- Celie, P.H.N.; Kasheverov, I.E.; Mordintsev, D.Y.; Hogg, R.C.; van Nierop, P.; van Elk, R.; van Rossum-Fikkert, S.E.; Zhmak, M.N.; Bertrand, D.; Tsetlin, V.; Sixma, T.K.; Smit, A.B. Crystal structure of nicotinic acetylcholine receptor homologue AChBP in complex with an α-conotoxin PnIA variant. Nature Struc. Mol. Biol. 2005, 12, 582–588. [Google Scholar]
- Hansen, S.B.; Sulzenbacher, G.; Huxford, T.; Marchot, P.; Taylor, P.; Bourne, Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005, 24, 3635–3646. [Google Scholar]
- Ulens, C.; Hogg, R.C.; Celie, P.H.; Bertrand, D.; Tsetlin, V.; Smit, A.B.; Sixma, T.K. Structural determinants of selective α-conotoxin binding to a nicotinic acetylcholine receptor homolog AChBP. Proc. Natl. Acad. Sci. USA 2006, 103, 3615–3620. [Google Scholar]
- Brejc, K.; van Dijk, W.J.; Klaasen, R.V.; Schuurmans, M.; van Der Oost, J.; Smit, A.B.; Sixma, T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411, 269–276. [Google Scholar]
- Hansen, S.B.; Talley, T.T.; Radic, Z.; Taylor, P. Structural and ligand recognition characteristics of an acetylcholine-binding protein from Aplysia californica. J. Biol. Chem. 2004, 279, 24197–24202. [Google Scholar]
- Celie, P.H.N.; Klaassen, R.V.; van Rossum-Fikkert, S.E.; van Elk, R.; van Nierop, P.; Smit, A.B.; Sixma, T.K. Crystal Structure of acetylcholine-binding Protein from Bulinus truncatus reveals the conserved structural scaffold and sites of variation in nicotinic acetylcholine receptors. J. Biol. Chem. 2005, 280, 26457–26466. [Google Scholar]
- Smit, A.B.; Syed, N.I.; Schaap, D.; van Minnen, J.; Klumperman, J.; Kits, K.S.; Lodder, H.; van der Schors, R.C.; Van Elk, R.; Sorgedrager, B.; Brejc, K.; Sixma, T.K.; Geraerts, W.P.M. A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature 2001, 411, 261–268. [Google Scholar]
- Celie, P.H.N.; Van Rossum-Fikkert, S.E.; Van Dijk, W.J.; Brejc, K.; Smit, A.B.; Sixma, T.K. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 2004, 41, 907–914. [Google Scholar]
- Le Novere, N.; Grutter, T.; Changeux, J.P. Models of the extracellular domain of the nicotinic receptors and of agonist and Ca2+ binding sites. Proc. Natl. Acad. Sci. USA 2002, 99, 3210–3215. [Google Scholar]
- Hansen, S.B.; Radic, Z.; Talley, T.T.; Molles, B.E.; Deerinick, T.; Tsigelny, I.; Taylor, P. Tryptophan fluorescence reveals conformational changes in the acetylcholine binding protein. J. Biol. Chem. 2002, 277, 41299–41302. [Google Scholar]
- Dutertre, S.; Lewis, R.J. Toxin insights into nicotinic acetylcholine receptors. Biochem. Pharmacol. 2006, 72, 661–670. [Google Scholar]
- Dellisanti, C.D.; Yao, Y.; Stroud, J.C.; Wang, Z.Z.; Chen, L. Crystal structure of the extracellular domain of nAChR alpha1 bound to alpha-bungarotoxin at 1.94 Å resolution. Nat. Neurosci. 2007, 10, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Hilf, R.J.C.; Dutzler, R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 2008, 452, 375–380. [Google Scholar]
- Bocquet, N.; Nury, H.; Baaden, M.; Le Poupon, C.; Changeux, J.P.; Delarue, M.; Corringer, P.-J. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature 2009, 457, 111–114. [Google Scholar]
- Hilf, R.J.C.; Dutzler, R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature 2009, 457, 115–119. [Google Scholar]
- Dutertre, S.; Lewis, R.J. Computational approaches to understand α-conotoxin interactions at neuronal nicotinic receptors. Eur. J. Biochem. 2004, 271, 2327–2334. [Google Scholar]
- Dutertre, S.; Nicke, A.; Tyndall, J.D.A.; Lewis, R.J. Determination of a-conotoxin binding modes on neuronal nicotinic acetylcholine receptors. J. Mol. Recognit. 2004, 17, 339–347. [Google Scholar]
- Pérez, E.M.; Cassels, B.K.; Zapata-Torres, G. Molecular modeling of the α9α10 nicotinic acetylcholine receptor subtype. Bioorg. Med. Chem. Lett. 2009, 19, 251–254. [Google Scholar]
- Westermann, J.C.; Clark, R.J.; Craik, D.J. Binding mode of α-conotoxins to an acetylcholine binding protein determined by saturation transfer difference NMR. Protein Pept. Lett. 2008, 15, 910–914. [Google Scholar]
- Ellison, M.; Gao, F.; Wang, H.L.; Sine, S.M.; McIntosh, J.M.; Olivera, B.M. α-Conotoxins ImI and ImII target distinct regions of the human alpha7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes. Biochemistry 2004, 43, 16019–16026. [Google Scholar]
- Servent, D.; Thanh, H.L.; Antil, S.; Bertrand, D.; Corringer, P.-J.; Changeux, J.P.; Menez, A. Functional determinants by which snake and cone snail toxins block the α7 neuronal nicotinic acetylcholine receptors. J. Physiol. Paris 1998, 92, 107–111. [Google Scholar]
- Rogers, J.P.; Luginbuhl, P.; Shen, G.S.; McCabe, R.T.; Stevens, R.C.; Wemmer, D.E. NMR solution structure of α-conotoxin ImI and comparison to other conotoxins specific for neuronal nicotinic acetylcholine receptors. Biochemistry 1999, 38, 3874–3882. [Google Scholar]
- Rogers, J.P.; Luginbuhl, P.; Pemberton, K.; Harty, P.; Wemmer, D.E.; Stevens, R.C. Structure-activity relationships in a peptidic α7 nicotinic acetylcholine receptor antagonist. J. Mol. Biol. 2000, 304, 911–926. [Google Scholar]
- Sine, S.M.; Bren, N.; Quiram, P.A. Molecular disection of subunit interfaces in the nicotinic acetylcholine receptor. J. Physiol. Paris 1998, 92, 101–105. [Google Scholar]
- Armishaw, C.; Jensen, A.A.; Balle, T.; Clark, R.J.; Harpsøe, K.; Skonberg, C.; Liljefors, T.; Strømgaard, K. The rational design of α-conotoxin analogues targeting α7 nicotinic acetylcholine receptors: Improved antagonistic activity by incorporation of proline derivatives. J. Biol. Chem. 2009, 284, 9498–9512. [Google Scholar]
- Kasheverov, I.E.; Zhmak, M.N.; Fish, A.; Rucktooa, P.; Khruschov, A.Y.; Osipov, A.V.; Ziganshin, R.H.; D'hoedt, D.; Bertrand, D.; Sixma, T.K.; Smit, A.B.; Tsetlin, V.I. Interactions of a-conotoxin ImII and its analogs with nicotinic receptors and acetylcholine binding proteins: additional binding sites on Torpedo receptor. J. Neurochem. 2009, 111, 934–944. [Google Scholar]
- Hogg, R.C.; Miranda, L.P.; Craik, D.J.; Lewis, R.J.; Alewood, P.F.; Adams, D.J. Single amino acid substitutions in α-conotoxin PnIA shift selectivity of the mammalian neuronal nicotinic acetyl choline receptor. J. Biol. Chem. 1999, 274, 36559–36564. [Google Scholar]
- Luo, S.; Nguyen, T.A.; Cartier, G.E.; Olivera, B.M.; Yoshikami, D.; McIntosh, J.M. Single residue alteration in α-conotoxin PnIA switches its nAChR subtype selectivity. Biochemistry 1999, 38, 14542–14548. [Google Scholar]
- Hogg, R.C.; Hopping, G.; Alewood, P.F.; Adams, D.J.; Bertrand, D. α-Conotoxins PnIA and [A10L]PnIA stabilize different states of the α7-L247T nicotinic acetylcholine receptor. J. Biol. Chem. 2003, 278, 26908–26914. [Google Scholar]
- Champtiaux, N.; Han, Z.-H.; Bessis, A.; Rossi, F.M.; Zoli, M.; Marubio, L.M.; McIntosh, J.M.; Changeux, J.P. Distribution and pharmacology of α6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J. Neurosci. 2002, 22, 1208–1217. [Google Scholar]
- McIntosh, J.M.; Azam, L.; Staheli, S.T.; Dowell, C.; Lindstrom, J.; Kuryatov, A.; Garret, J.E.; Marks, M.J.; Whiteaker, P. Analogs of α-conotoxin MII are selective for α6-containing nicotinic acetylcholine receptors. Mol. Pharmacol. 2004, 65, 944–952. [Google Scholar]
- Bordia, T.; Grady, S.R.; McIntosh, J.M.; Quik, M. Nigrostriatal damage preferentially decreases a subpopulation of α6β2* nAChRs in mouse, monkey, and Parkinson's disease striatum. Mol. Pharmacol. 2007, 72, 52–61. [Google Scholar]
- Meyer, E.L.; Yoshikami, D.; McIntosh, J.M. The neuronal nicotinic acetylcholine receptors α4* and α6* differentially modulate dopamine release in mouse striatal slices. J. Neurochem. 2008, 105, 1761–1769. [Google Scholar]
- Turner, M.; Eidemiller, S.; Martin, B.; Narver, A.; Marshall, J.; Zemp, L.; Cornell, K.A.; McIntosh, J.M.; McDougal, O.M. Structural basis for α-conotoxin potency and selectivity. Bioorgan. Med. Chem. 2009, 17, 5894–5899. [Google Scholar]
- Azam, L.; Yoshikami, D.; McIntosh, J.M. Amino acid residues that confer high selectivity of the alpha 6 nicotinic acetylcholine receptor subunit to alpha -conotoxin MII [S4A,E11A,L15A]. J. Biol. Chem. 2008, 283, 11625–11632. [Google Scholar]
- Millard, E.L.; Nevin, S.T.; Loughnan, M.L.; Nicke, A.; Clark, R.J.; Alewood, P.F.; Lewis, R.J.; Adams, D.J.; Craik, D.J.; Daly, N.L. Inhibition of neuronal nicotinic acetylcholine receptor subtypes by α-conotoxin GID and analogues. J. Biol. Chem. 2009, 284, 4944–4951. [Google Scholar]
- Whiteaker, P.; Marks, M.J.; Christensen, S.; Dowell, C.; Collins, A.C.; McIntosh, J.M. Synthesis and characterization of [125I]α-conotoxin ArIB[V11L;V16A], a selective α7 nAChR antagonist. J. Pharmacol. Exp. Ther. 2008, 325, 910–919. [Google Scholar]
- Vincler, M.; McIntosh, J.M. Targeting the α9α10 nicotinic acetylcholine receptor to treat severe pain. Expert Opin. Ther. Targets 2007, 11, 891–897. [Google Scholar]
- Nevin, S.T.; Clark, R.J.; Klimis, H.; Christie, M.J.; Craik, D.J.; Adams, D.J. Are α9α10 nicotinic acetylcholine receptors a pain target for α-conotoxins? Mol. Pharmacol. 2007, 72, 1406–1410. [Google Scholar] [CrossRef] [PubMed]
- Satkunanathan, N.; Livett, B.; Gayler, K.; Sandall, D.; Down, J.; Khalil, Z. α-Conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005, 1059, 149–158. [Google Scholar]
- Vincler, M.; Wittenauer, S.; Parker, R.; Ellison, M.; Olivera, B.M.; McIntosh, J.M. Molecular mechanisms for analgesia involving specific antagonism of α9α10 nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. USA 2006, 103, 17880–17884. [Google Scholar]
- McIntosh, J.M.; Absalom, N.; Chebib, M.; Elgoyhen, A.B.; Vincler, M. Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem. Pharmacol. 2009, 78, 693–702. [Google Scholar]
- Clark, R.J.; Fischer, H.; Nevin, S.T.; Adams, D.J.; Craik, D.J. The synthesis, structural characterization, and receptor specificity of the α-conotoxin Vc1.1. J. Biol. Chem. 2006, 281, 23254–23263. [Google Scholar]
- Callaghan, B.; Haythornthwaite, A.; Berecki, G.; Clark, R.J.; Craik, D.J.; Adams, D.J. Analgesic alpha-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J. Neurosci. 2008, 28, 10943–10951. [Google Scholar] [CrossRef] [PubMed]
- Halai, R.; Clark, R.J.; Nevin, S.T.; Jensen, J.E.; Adams, D.J.; Craik, D.J. Scanning mutagenisis of α-conotoxin Vc1.1 reveals residues crucial for activity at the α9α10 nicotinic acetylcholine receptor. J. Biol. Chem. 2009, 284, 20275–20284. [Google Scholar] [PubMed]
- Ellison, M.; Feng, Z.-P.; Park, A.J.; Zhang, X.; Olivera, B.M.; McIntosh, J.M.; Norton, R.S. α-RgIA, a novel conotoxin that blocks the α9α10 nAChR: Structure and identification of key receptor-binding residues. J. Mol. Biol. 2008, 377, 1216–1227. [Google Scholar]
- Alewood, P.; Hopping, G.; Armishaw, C. Marine Toxins as a Source of Drug Leads. Aust. J. Chem. 2003, 56, 769–774. [Google Scholar]
- Bulaj, G.; Olivera, B.M. Folding of conotoxins: Formation of the native disulfide bridges during chemical synthesis and biosynthesis of Conus peptides. Antioxid. Redox Signal. 2008, 10, 141–155. [Google Scholar]
- Brust, A.; Tickle, A.E. High-throughput synthesis of conopeptides: a safety-catch linker approach enabling disulfide formation in 96-well format. J. Peptide Sci. 2007, 13, 133–141. [Google Scholar]
- Patek, M.; Lebl, M. A Safety-Catch Type of Amide Protecting Group. Tetrahedron Lett. 1990, 31, 5209–5212. [Google Scholar]
- Muttenthaler, M.; Nevin, S.T.; Grishin, A.A.; Ngo, S.T.; Choy, P.T.; Daly, N.L.; Hu, S.-H.; Armishaw, C.J.; Wang, C.-I.; Lewis, R.J.; Martin, J.L.; Noakes, P.G.; Craik, D.J.; Adams, D.J.; Alewood, P.F. Solving the α-conotoxin folding problem: Efficient selenium-directed on-resin generation of more potent and stable nicotinic acetylcholine receptor antagonists. J. Am. Chem. Soc. 2010, 132, 3514–3522. [Google Scholar]
- Armishaw, C.J.; Singh, N.; Medina-Franco, J.; Clark, R.J.; Scott, K.C.M.; Houghten, R.A.; Jensen, A.A. A synthetic combinatorial strategy for developing α-conotoxin analogs as potent α7 nicotinic acetylcholine receptor antagonists. J. Biol. Chem. 2010, 285, 1809–1821. [Google Scholar]
- Terlau, H.; Olivera, B.M. Conus Venoms: A Rich Source of Novel Ion Channel-Targeted Peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar]
- Houghten, R.A. Parallel array and mixture-based synthetic combinatorial chemistry. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 273–282. [Google Scholar]
- Pinilla, C.; Appel, J.R.; Blanc, P.; Houghten, R.A. Rapid identification of high affinity peptide ligands using positional scanning synthetic peptide combinatorial libraries. Biotechniques 1992, 13, 901–905. [Google Scholar]
- Dooley, C.T.; Ny, P.; Bidlack, J.M.; Houghten, R.A. Selective ligands for the μ, δ and κ opioid receptors identifies from a single mixture based tetrapeptide positional scan combinatorial library. J. Biol. Chem. 1998, 273, 18848–18856. [Google Scholar]
- Pinilla, C.; Rubio-Godoy, V.; Dutoit, V.; Guillaume, P.; Simon, R.; Zhao, Y.; Houghten, R.A.; Cerottini, J.C.; Romero, P.; Valmori, D. Combinatorial peptide libraries as an alternative approach to the identification of ligands for tumor-reactive cytolytic T lymphocytes. Cancer Res. 2001, 61, 5153–5160. [Google Scholar]
- Rubio-Godoy, V.; Dutoit, V.; Zhao, Y.; Simon, R.; Guillaume, P.; Houghten, R.A.; Romero, P.; Cerottini, J.C.; Pinilla, C.; Valmori, D. Positional scanning-synthetic peptide library-based analysis of self- and pathogen-derived peptide cross-reactivity with tumor-reactive Melan-A-specific CTL. J. Immunol. 2002, 169, 5696–5707. [Google Scholar]
- Bersanetti, P.A.; Andrade, M.C.; Casarini, D.E.; Juliano, M.A.; Nchinda, A.T.; Sturrock, E.D.; Juliano, L.; Carmona, A.K. Positional-scanning combinatorial libraries of fluorescence resonance energy transfer peptides for defining substrate specificity of the angiotensin I-converting enzyme and development of selective C-domain substrates. Biochemistry 2004, 43, 15729–15736. [Google Scholar]
- Kopecky, E.M.; Greinstetter, S.; Pabinger, I.; Buchacher, A.; Romisch, J.; Jungbauer, A. Combinatorial peptides direcetd to inhibitory antibodies against human blood clotting factor VIII. Thromb. Haemost. 2005, 94, 933–941. [Google Scholar]
- Ryge, T.S.; Hansen, P.R. Potent antibacterial lysine-peptoid hybrids identified from a positional scanning combinatorial library. Bioorgan. Med. Chem. 2006, 14, 4444–4451. [Google Scholar]
- Denholt, C.L.; Hansen, P.R.; Pedersen, N.; Poulsen, H.S.; Gillings, N.; Kjaer, A. Identification of novel peptide ligands for the cancer-specific receptor mutation EFGRvIII using a mixture based synthetic combinatorial library. Biopolymers 2009, 91, 201–206. [Google Scholar]
- Schneider, E.L.; Craik, C.S. Positional scanning synthetic combinatorial libraries for substrate profiling. Meth. Mol. B 2009, 539, 59–78. [Google Scholar]
- Kessler, H. Conformation and biological activity of cyclic peptides. Angew. Chem. Int. Ed. 1982, 21, 512–523. [Google Scholar]
- Clark, R.J.; Fischer, H.; Dempster, L.; Daly, N.L.; Rosengren, K.J.; Nevin, S.T.; Meunier, F.A.; Adams, D.J.; Craik, D.J. Engineering stable peptide toxins by means of backbone cyclization: Stabilization of the α-conotoxin MII. Proc. Natl. Acad. Sci. USA 2005, 102, 13767–13772. [Google Scholar]
- Lovelace, E.S.; Armishaw, C.J.; Colgrave, M.L.; Wahlstrom, M.E.; Alewood, P.F.; Daly, N.L.; Craik, D.J. Cyclic MrIA: A stable and potent cyclic conotoxin with a novel topological fold that targets the norepinephrine transporter. J. Med. Chem. 2006, 49, 6561–6568. [Google Scholar]
- Armishaw, C.J.; Dutton, J.L.; Craik, D.J.; Alewood, P.F. Establishing regiocontrol of disulfide bond isomers of α-conotoxin ImI via the synthesis of N-to-C cyclic analogues. Pept. Sci. 2010, 94, 307–313. [Google Scholar]
- Blanchfield, J.T.; Dutton, J.L.; Hogg, R.C.; Gallagher, O.P.; Craik, D.J.; Jones, A.; Adams, D.J.; Lewis, R.J.; Alewood, P.F.; Toth, I. Synthesis, structure elucidation, in vitro biological activity, toxicity, and caco-2 cell permeability of lipophilic analogues of α-conotoxin MII. J. Med. Chem. 2003, 46, 1266–1272. [Google Scholar]
- Blanchfield, J.T.; Gallagher, O.P.; Cros, C.; Lewis, R.J.; Alewood, P.F.; Toth, I. Oral absorption and in vivo biodistribution of α-conotoxin MII and a lipidic analogue. Biochem. Biophys. Res. Commun. 2007, 361, 97–102. [Google Scholar]
- Dekan, Z.; Paczkowski, F.A.; Lewis, R.J.; Alewood, P.F. Synthesis and in Vitro Biological Activity of Cyclic Lipophilic χ-Conotoxin MrIA Analogues. Int. J. Pept. Res. Ther. 2007, 13, 307–312. [Google Scholar]
- Hargittai, B.; Solé, N.A.; Groebe, D.R.; Abramson, S.N.; Barany, G. Chemical synthesis and biological activities of lactam analogues of α-conotoxin SI. J. Med. Chem. 2000, 43, 4787–4792. [Google Scholar]
- Bondebjerg, J.; Grunnet, M.; Jespersen, T.; Meldal, M. Solid-phase synthesis and biological activity of a thioether analogue of conotoxin GI. Chembiochem 2003, 4, 186–194. [Google Scholar]
- MacRaild, C.A.; Illesinghe, J.; van Lierop, B.J.; Townsend, A.L.; Chebib, M.; Livett, B.G.; Robinson, A.J.; Norton, R.S. Structure and activity of (2,8)-Dicarba-(3,12)-cystino α-ImI, an α-conotoxin containing a nonreducible cystine analogue. J. Med. Chem. 2009, 52, 755–762. [Google Scholar]
- Armishaw, C.J.; Daly, N.L.; Nevin, S.T.; Adams, D.J.; Craik, D.J.; Alewood, P.F. α-Selenoconotoxins: A new class of potent α7 neuronal nicotinic receptor antagonists. J. Biol. Chem. 2006, 281, 14136–14143. [Google Scholar]
- Walewska, A.; Zhang, M.-M.; Skalicky, J.J.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Integrated oxidative folding of cysteine/selenocysteine containing peptides: improving chemical synthesis of conotoxins. Angew. Chem. Int. Ed. 2009, 48, 2221–2224. [Google Scholar]
- Gowd, K.H.; Yarotskyy, V.; Elmslie, K.S.; Skalicky, J.J.; Olivera, B.M.; Bulaj, G. Site-specific effects of diselenide bridges on the oxidative folding of a cystine knot peptide, ω-conotoxin GVIA. Biochemistry 2010, 49, 2741–2752. [Google Scholar]
- Besse, D.; Moroder, L. Synthesis of selenocysteine peptides and their oxidation to diselenide-bridged compounds. J. Pept. Sci. 1997, 3, 442–453. [Google Scholar]
- Muttenthaler, M.; Alewood, P.F. Selenopeptide chemistry. J. Pept. Sci. 2008, 14, 1223–1239. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Armishaw, C.J. Synthetic α-Conotoxin Mutants as Probes for Studying Nicotinic Acetylcholine Receptors and in the Development of Novel Drug Leads. Toxins 2010, 2, 1471-1499. https://doi.org/10.3390/toxins2061471
Armishaw CJ. Synthetic α-Conotoxin Mutants as Probes for Studying Nicotinic Acetylcholine Receptors and in the Development of Novel Drug Leads. Toxins. 2010; 2(6):1471-1499. https://doi.org/10.3390/toxins2061471
Chicago/Turabian StyleArmishaw, Christopher J. 2010. "Synthetic α-Conotoxin Mutants as Probes for Studying Nicotinic Acetylcholine Receptors and in the Development of Novel Drug Leads" Toxins 2, no. 6: 1471-1499. https://doi.org/10.3390/toxins2061471