The Glucocorticoid Receptor: A Revisited Target for Toxins

Abstract
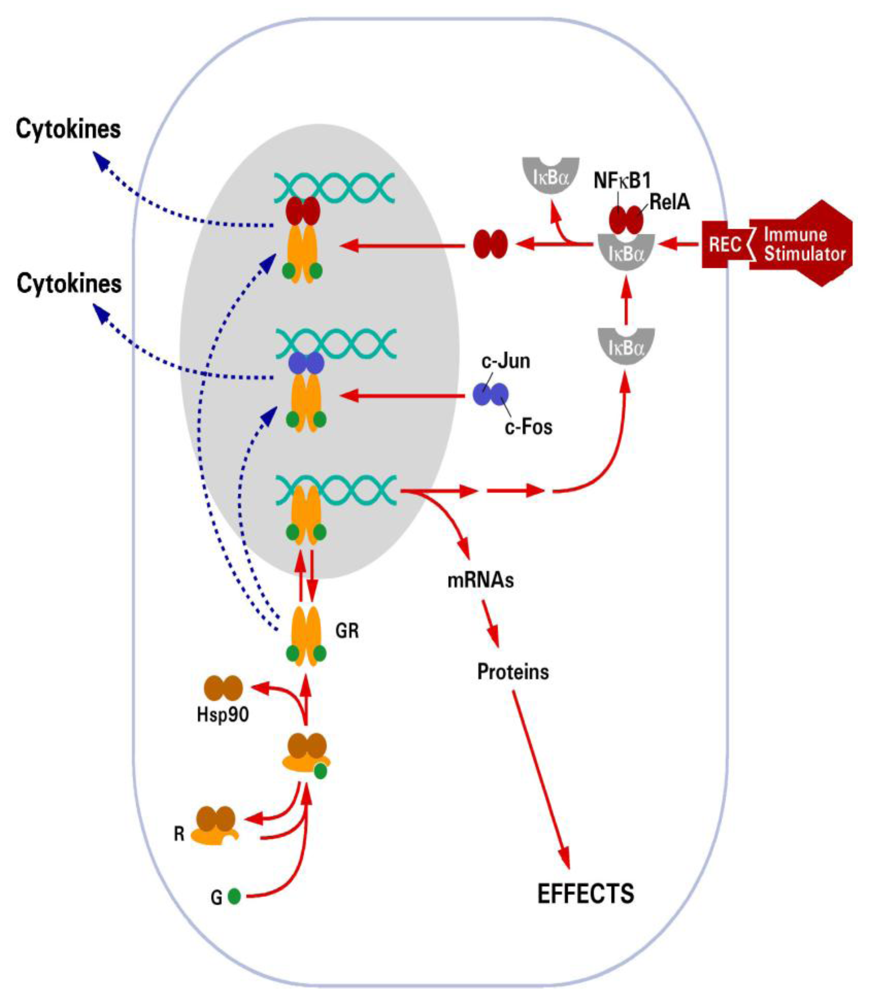
:1. Introduction
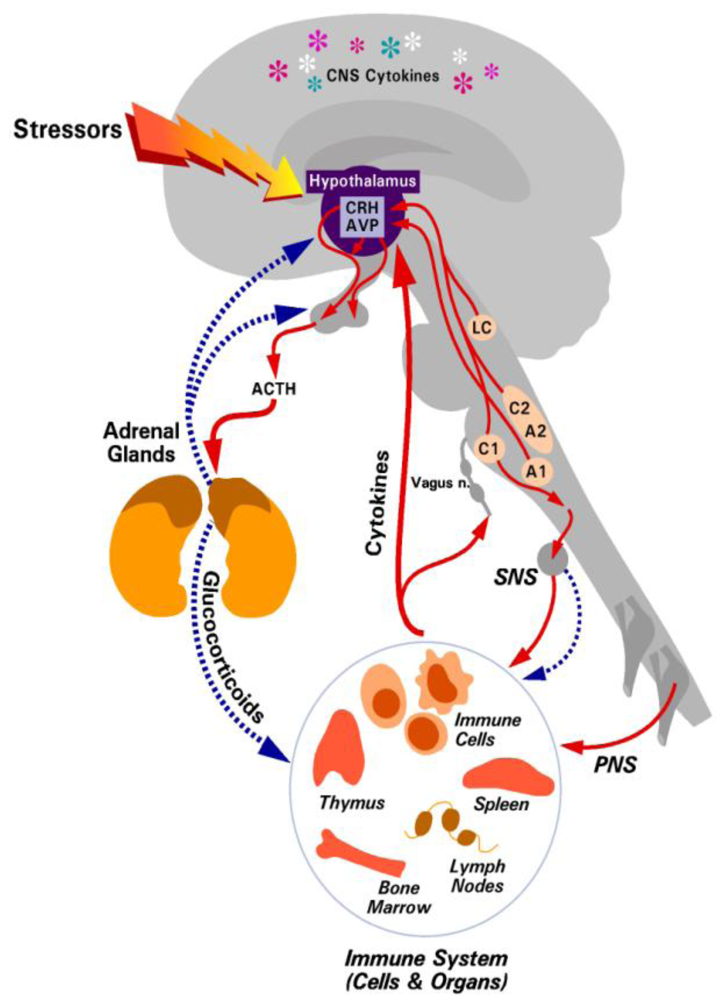
2. The Hypothalamic-Pituitary-Adrenal (HPA) Axis and Glucocorticoid Responses
2.1. Disruption of the HPA Axis/Glucocorticoid Responses Increases Mortality

2.2. Glucocorticoid Receptor (GR)

3. Effect of Bacterial Toxins on GR

{kind=link}
{kind=link}
| Toxin | Effect on GR | Reference |
|---|---|---|
| Aflatoxin B1 | Decreases glucocorticoid induction of liver ribonucleic acid synthesis | [61] |
| Decreases nuclear GR ligand binding | [62,63] | |
| Decreases glucocorticoid induction of liver enzymes | [64,65] | |
| Anthrax lethal toxin | Represses GR-mediated gene activation | [66,67] |
| Clostridial toxins | Represses GR-induced gene activation | [68] |
| Prevents glucocorticoid repression of cytokine production | [68] | |
| Endotoxin/LPS | Impairs glucocorticoid regulation of liver enzymes | [69,70,71,72] |
| Decreases GR ligand binding | [11,70,71,72,73,74,75] | |
| Decreases GR numbers and affinity in lungs | [76] | |
| Increases GR numbers but decreases affinity in bronchial epithelial cell line | [77] | |
| Reduces glucocorticoid induction of GR responsive promoter in cell culture | [78,79,80] | |
| Increases GR numbers in murine macrophages | [81] | |
| No effect on hepatic GR numbers or affinity | [82] | |
| Shiga toxin | Increases GR numbers in neutrophils | [17] |
| Superantigen | Induces glucocorticoid resistance | [83,84,85] |
| Impairs GR nuclear translocation | [84] | |
| Induces GRβ | [83,86,87] |
3.1. Anthrax Lethal Toxin
3.2. Endotoxin/LPS
3.3. Shiga Toxin
3.4. Bacterial Superantigens
3.5. Clostridia Toxins
4. Effect of Mycotoxins and Plant Toxins on GR
5. Effect of Environmental and Chemical Toxins on GR
| Toxin | Effect on GR | Reference |
|---|---|---|
| Arsenic | Low dose represses GR-mediated gene activation | [116,117,118,119,120] |
| Inhibits GR ligand binding | [121,122,123,124,125] | |
| Extreme low dose enhances GR-mediated gene activation | [117,118,119] | |
| Reduces CARM1 binding to GR-regulated promoter | [116] | |
| Beryllium | Inhibits glucocorticoid induction of liver enzymes | [126,127] |
| Cadmium | Low dose reduces GR-mediated gene activation | [121,128] |
| High dose enhances GR-mediated activation | [121] | |
| Inhibits GR ligand binding in liver | [121,124] | |
| Inhibits GR DNA binding in liver | [121] | |
| Chromium | Extreme low dose enhances GC-induced liver enzymes | [119,129] |
| Decreases glucocorticoid-induced liver genes | [119,129] | |
| Lead | Inhibits glucocorticoid induction of liver genes | [130] |
| Mercury | Reduces glucocorticoid induction of liver genes | [131] |
| Decreases GR ligand binding | [132] | |
| Enhances interaction between GR and Hsp proteins | [133] | |
| Enhances GR-responsive MMTV promoter | [134] | |
| Selenite | Inhibits GR ligand binding | [123,135] |
| Decreases glucocorticoid induction of GR-regulated genes | [85] | |
| Zinc | Reduces GR ligand binding in liver | [136] |
| Enhances GR-responsive MMTV promoter | [134] |
| Effect on GR | Reference |
|---|---|
| Reduces GR ligand binding affinity in bronchial epithelial cells | [137] |
| No difference in GR mRNA levels in bronchial epithelial cells | [138] |
| Reduces GRα protein levels in mouse lungs exposed to cigarette smoke | [139] |
| No difference in GRα/β mRNA levels in bronchial epithelial cells | [138] |
| Reduces GR α/β protein levels in PBMCs | [140] |
| Reduces CYP3A5 expression in alveolar macrophages | [141] |
| Inhibits glucocorticoid-induction of ENaC mRNA | [142] |
| Inhibits glucocorticoid repression of cytokine production in BAL macrophages | [143] |
| Inhibits HDAC2 expression and activity | [143] |
5.1. Heavy Metals
5.2. Cigarette Smoke
6. Effect of Toxins on Other Nuclear Hormone Receptors
7. Clinical Relevance
8. Conclusions
Acknowledgements
References
- Pessini, A.C.; de Souza, A.M.; Faccioli, L.H.; Gregorio, Z.M.; Arantes, E.C. Time course of acute-phase response induced by Tityus serrulatus venom and TsTX-I in mice. Int. Immunopharmacol. 2003, 3, 765–774. [Google Scholar]
- Barros, S.F.; Friedlanskaia, I.; Petricevich, V.L.; Kipnis, T.L. Local inflammation, lethality and cytokine release in mice injected with Bothrops atrox venom. Mediat. Inflamm. 1998, 7, 339–346. [Google Scholar]
- Lu, J.; Wang, A.; Ansari, S.; Hershberg, R.M.; McKay, D.M. Colonic bacterial superantigens evoke an inflammatory response and exaggerate disease in mice recovering from colitis. Gastroenterology 2003, 125, 1785–1795. [Google Scholar]
- Teixeira Cde, F.; Fernandes, C.M.; Zuliani, J.P.; Zamuner, S.F. Inflammatory effects of snake venom metalloproteinases. Mem. Inst. Oswaldo Cruz 2005, 100, 181–184. [Google Scholar]
- Berczi, I. Neurohormonal host defense in endotoxin shock. Ann. N. Y. Acad. Sci. 1998, 840, 787–802. [Google Scholar]
- Haddad, J.J. On the mechanisms and putative pathways involving neuroimmune interactions. Biochem. Biophys. Res. Commun. 2008, 370, 531–535. [Google Scholar]
- Marques-Deak, A.; Cizza, G.; Sternberg, E. Brain-immune interactions and disease susceptibility. Mol. Psychiatr. 2005, 10, 239–250. [Google Scholar]
- Webster, J.I.; Tonelli, L.; Sternberg, E.M. Neuroendocrine regulation of immunity. Annu. Rev. Immunol. 2002, 20, 125–163. [Google Scholar]
- Bellinger, D.L.; Millar, B.A.; Perez, S.; Carter, J.; Wood, C.; ThyagaRajan, S.; Molinaro, C.; Lubahn, C.; Lorton, D. Sympathetic modulation of immunity: Relevance to disease. Cell. Immunol. 2008, 252, 27–56. [Google Scholar]
- Rosas-Ballina, M.; Tracey, K.J. The neurology of the immune system: Neural reflexes regulate immunity. Neuron 2009, 64, 28–32. [Google Scholar]
- McCallum, R.E.; Stith, R.D. Endotoxin-induced inhibition of steroid binding by mouse liver cytosol. Circ. Shock 1982, 9, 357–367. [Google Scholar]
- Morrow, L.E.; McClellan, J.L.; Conn, C.A.; Kluger, M.J. Glucocorticoids alter fever and IL-6 responses to psychological stress and to lipopolysaccharide. Am. J. Physiol. 1993, 264, R1010–R1016. [Google Scholar]
- Butler, L.D.; Layman, N.K.; Riedl, P.E.; Cain, R.L.; Shellhaas, J.; Evans, G.F.; Zuckerman, S.H. Neuroendocrine regulation of in vivo cytokine production and effects: I. In vivo regulatory networks involving the neuroendocrine system, interleukin-1 and tumor necrosis factor-alpha. J. Neuroimmunol. 1989, 24, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.M.; Souza, G.E.; Pela, I.R. Endotoxin-induced fever is modulated by endogenous glucocorticoids in rats. Am. J. Physiol. 1992, 263, R423–R427. [Google Scholar]
- Nakano, K.; Suzuki, S.; Oh, C. Significance of increased secretion of glucocorticoids in mice and rats injected with bacterial endotoxin. Brain Behav. Immun. 1987, 1, 159–172. [Google Scholar]
- Silverstein, R.; Hannah, P.; Johnson, D.C. Natural adrenocorticosteroids do not restore resistance to endotoxin in the adrenalectomized mouse. Circ. Shock 1993, 41, 162–165. [Google Scholar]
- Gomez, S.A.; Fernandez, G.C.; Vanzulli, S.; Dran, G.; Rubel, C.; Berki, T.; Isturiz, M.A.; Palermo, M.S. Endogenous glucocorticoids attenuate Shiga toxin-2-induced toxicity in a mouse model of haemolytic uraemic syndrome. Clin. Exp. Immunol. 2003, 131, 217–224. [Google Scholar]
- Gonzalo, J.A.; Gonzalez-Garcia, A.; Kalland, T.; Hedlund, G.; Martinez, C.; Kroemer, G. Linomide, a novel immunomodulator that prevents death in four models of septic shock. Eur. J. Immunol. 1993, 23, 2372–2374. [Google Scholar]
- Sternberg, E.M.; Hill, J.M.; Chrousos, G.P.; Kamilaris, T.; Listwak, S.J.; Gold, P.W.; Wilder, R.L. Inflammatory mediator-induced hypothalamic-pituitary-adrenal axis activation is defective in streptococcal cell wall arthritis-susceptible Lewis rats. Proc. Natl. Acad. Sci. USA 1989, 86, 2374–2378. [Google Scholar]
- Castagliuolo, I.; Karalis, K.; Valenick, L.; Pasha, A.; Nikulasson, S.; Wlk, M.; Pothoulakis, C. Endogenous corticosteroids modulate Clostridium difficile toxin A-induced enteritis in rats. Amer. J. Physiol.-Gastrointest L. 2001, 280, G539–G545. [Google Scholar]
- Mykoniatis, A.; Anton, P.M.; Wlk, M.; Wang, C.C.; Ungsunan, L.; Bluher, S.; Venihaki, M.; Simeonidis, S.; Zacks, J.; Zhao, D.; Sougioultzis, S.; Karalis, K.; Mantzoros, C.; Pothoulakis, C. Leptin mediates Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology 2003, 124, 683–691. [Google Scholar]
- Palermo, M.; Alves-Rosa, F.; Rubel, C.; Fernandez, G.C.; Fernandez-Alonso, G.; Alberto, F.; Rivas, M.; Isturiz, M. Pretreatment of mice with lipopolysaccharide (LPS) or IL-1beta exerts dose-dependent opposite effects on Shiga toxin-2 lethality. Clin. Exp. Immunol. 2000, 119, 77–83. [Google Scholar]
- Lazar, G., Jr.; Duda, E.; Lazar, G. Effect of RU 38486 on TNF production and toxicity. FEBS Lett. 1992, 308, 137–140. [Google Scholar]
- Hawes, A.S.; Rock, C.S.; Keogh, C.V.; Lowry, S.F.; Calvano, S.E. In vivo effects of the antiglucocorticoid RU 486 on glucocorticoid and cytokine responses to Escherichia coli endotoxin. Infect. Immun. 1992, 60, 2641–2647. [Google Scholar]
- Reichardt, H.M.; Umland, T.; Bauer, A.; Kretz, O.; Schutz, G. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol. Cell. Biol. 2000, 20, 9009–9017. [Google Scholar]
- Moayeri, M.; Webster, J.I.; Wiggins, J.F.; Leppla, S.H.; Sternberg, E.M. Endocrine perturbation increases susceptibility of mice to anthrax lethal toxin. Infect. Immun. 2005, 73, 4238–4244. [Google Scholar]
- Lu, N.Z.; Wardell, S.E.; Burnstein, K.L.; Defranco, D.; Fuller, P.J.; Giguere, V.; Hochberg, R.B.; McKay, L.; Renoir, J.M.; Weigel, N.L.; Wilson, E.M.; McDonnell, D.P.; Cidlowski, J.A. International Union of Pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: Glucocorticoid, mineralocorticoid, progesterone, and androgen receptors. Pharmacol. Rev. 2006, 58, 782–797. [Google Scholar] [CrossRef] [PubMed]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar]
- Jantzen, H.M.; Strahle, U.; Gloss, B.; Stewart, F.; Schmid, W.; Boshart, M.; Miksicek, R.; Schutz, G. Cooperativity of glucocorticoid response elements located far upstream of the tyrosine aminotransferase gene. Cell 1987, 49, 29–38. [Google Scholar]
- Drouin, J.; Trifiro, M.A.; Plante, R.K.; Nemer, M.; Eriksson, P.; Wrange, O. Glucocorticoid receptor binding to a specific DNA sequence is required for hormone-dependent repression of pro-opiomelanocortin gene transcription. Mol. Cell. Biol. 1989, 9, 5305–5314. [Google Scholar]
- Kassel, O.; Herrlich, P. Crosstalk between the glucocorticoid receptor and other transcription factors: Molecular aspects. Mol. Cell. Endocrinol. 2007, 275, 13–29. [Google Scholar]
- Karin, M.; Chang, L. AP-1-glucocorticoid receptor crosstalk taken to a higher level. J. Endocrinol. 2001, 169, 447–451. [Google Scholar]
- Cole, T.J.; Blendy, J.A.; Monaghan, A.P.; Krieglstein, K.; Schmid, W.; Aguzzi, A.; Fantuzzi, G.; Hummler, E.; Unsicker, K.; Schutz, G. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995, 9, 1608–1621. [Google Scholar]
- Reichardt, H.M.; Kaestner, K.H.; Tuckermann, J.; Kretz, O.; Wessely, O.; Bock, R.; Gass, P.; Schmid, W.; Herrlich, P.; Angel, P.; Schutz, G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 1998, 93, 531–541. [Google Scholar]
- Charmandari, E.; Kino, T.; Souvatzoglou, E.; Vottero, A.; Bhattacharyya, N.; Chrousos, G.P. Natural glucocorticoid receptor mutants causing generalized glucocorticoid resistance: Molecular genotype, genetic transmission, and clinical phenotype. J. Clin. Endocrinol. Metab. 2004, 89, 1939–1949. [Google Scholar]
- Leung, D.Y.; Spahn, J.D.; Szefler, S.J. Steroid-unresponsive asthma. Semin. Respir. Crit. Care Med. 2002, 23, 387–398. [Google Scholar]
- Kojika, S.; Sugita, K.; Inukai, T.; Saito, M.; Iijima, K.; Tezuka, T.; Goi, K.; Shiraishi, K.; Mori, T.; Okazaki, T.; Kagami, K.; Ohyama, K.; Nakazawa, S. Mechanisms of glucocorticoid resistance in human leukemic cells: implication of abnormal 90 and 70 kDa heat shock proteins. Leukemia 1996, 10, 994–999. [Google Scholar]
- Matysiak, M.; Makosa, B.; Walczak, A.; Selmaj, K. Patients with multiple sclerosis resisted to glucocorticoid therapy: Abnormal expression of heat-shock protein 90 in glucocorticoid receptor complex. Mult. Scler. 2008, 14, 919–926. [Google Scholar]
- Ouyang, J.; Jiang, T.; Tan, M.; Cui, Y.; Li, X. Abnormal expression and distribution of heat shock protein 90: Potential etiologic immunoendocrine mechanism of glucocorticoid resistance in idiopathic nephrotic syndrome. Clin. Vaccine Immunol. 2006, 13, 496–500. [Google Scholar]
- Qian, X.; Zhu, Y.; Xu, W.; Lin, Y. Glucocorticoid receptor and heat shock protein 90 in peripheral blood mononuclear cells from asthmatics. Chin. Med. J. 2001, 114, 1051–1054. [Google Scholar]
- Koga, Y.; Matsuzaki, A.; Suminoe, A.; Hattori, H.; Kanemitsu, S.; Hara, T. Differential mRNA expression of glucocorticoid receptor alpha and beta is associated with glucocorticoid sensitivity of acute lymphoblastic leukemia in children. Pediatr. Blood Cancer 2005, 45, 121–127. [Google Scholar]
- Lewis-Tuffin, L.J.; Cidlowski, J.A. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann. N. Y. Acad. Sci. 2006, 1069, 1–9. [Google Scholar]
- Pujols, L.; Mullol, J.; Picado, C. Alpha and beta glucocorticoid receptors: Relevance in airway diseases. Curr. Allergy Asthma Rep. 2007, 7, 93–99. [Google Scholar]
- Pujols, L.; Xaubet, A.; Ramirez, J.; Mullol, J.; Roca-Ferrer, J.; Torrego, A.; Cidlowski, J.A.; Picado, C. Expression of glucocorticoid receptors alpha and beta in steroid sensitive and steroid insensitive interstitial lung diseases. Thorax 2004, 59, 687–693. [Google Scholar]
- Towers, R.; Naftali, T.; Gabay, G.; Carlebach, M.; Klein, A.; Novis, B. High levels of glucocorticoid receptors in patients with active Crohn's disease may predict steroid resistance. Clin. Exp. Immunol. 2005, 141, 357–362. [Google Scholar]
- Pretorius, E.; Wallner, B.; Marx, J. Cortisol resistance in conditions such as asthma and the involvement of 11beta-HSD-2: A hypothesis. Horm. Metab. Res. 2006, 38, 368–376. [Google Scholar]
- Irusen, E.; Matthews, J.G.; Takahashi, A.; Barnes, P.J.; Chung, K.F.; Adcock, I.M. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: Role in steroid-insensitive asthma. J. Allergy Clin. Immunol. 2002, 109, 649–657. [Google Scholar]
- Matthews, J.G.; Ito, K.; Barnes, P.J.; Adcock, I.M. Defective glucocorticoid receptor nuclear translocation and altered histone acetylation patterns in glucocorticoid-resistant patients. J. Allergy Clin. Immunol. 2004, 113, 1100–1108. [Google Scholar]
- Tao, T.; Lan, J.; Lukacs, G.L.; Hache, R.J.; Kaplan, F. Importin 13 regulates nuclear import of the glucocorticoid receptor in airway epithelial cells. Am. J. Respir. Cell. Mol. Biol. 2006, 35, 668–680. [Google Scholar]
- Chrousos, G.P. A new "new" syndrome in the new world: Is multiple postreceptor steroid hormone resistance due to a coregulator defect? J. Clin. Endocrinol. Metab. 1999, 84, 4450–4453. [Google Scholar]
- New, M.I.; Nimkarn, S.; Brandon, D.D.; Cunningham-Rundles, S.; Wilson, R.C.; Newfield, R.S.; Vandermeulen, J.; Barron, N.; Russo, C.; Loriaux, D.L.; O'Malley, B. Resistance to multiple steroids in two sisters. J. Steroid Biochem. Mol. Biol. 2001, 76, 161–166. [Google Scholar]
- Diaz-Borjon, A.; Richaud-Patin, Y.; Alvarado de la Barrera, C.; Jakez-Ocampo, J.; Ruiz-Arguelles, A.; Llorente, L. Multidrug resistance-1 (MDR-1) in rheumatic autoimmune disorders. Part II: Increased P-glycoprotein activity in lymphocytes from systemic lupus erythematosus patients might affect steroid requirements for disease control. Joint Bone Spine 2000, 67, 40–48. [Google Scholar] [PubMed]
- Farrell, R.J.; Kelleher, D. Glucocorticoid resistance in inflammatory bowel disease. J. Endocrinol. 2003, 178, 339–346. [Google Scholar]
- Farrell, R.J.; Murphy, A.; Long, A.; Donnelly, S.; Cherikuri, A.; O'Toole, D.; Mahmud, N.; Keeling, P.W.; Weir, D.G.; Kelleher, D. High multidrug resistance (P-glycoprotein 170) expression in inflammatory bowel disease patients who fail medical therapy. Gastroenterology 2000, 118, 279–288. [Google Scholar]
- Hirano, T.; Onda, K.; Toma, T.; Miyaoka, M.; Moriyasu, F.; Oka, K. MDR1 mRNA expressions in peripheral blood mononuclear cells of patients with ulcerative colitis in relation to glucocorticoid administration. J. Clin. Pharmacol. 2004, 44, 481–486. [Google Scholar]
- Llorente, L.; Richaud-Patin, Y.; Diaz-Borjon, A.; Alvarado de la Barrera, C.; Jakez-Ocampo, J.; de la Fuente, H.; Gonzalez-Amaro, R.; Diaz-Jouanen, E. Multidrug resistance-1 (MDR-1) in rheumatic autoimmune disorders. Part I: Increased P-glycoprotein activity in lymphocytes from rheumatoid arthritis patients might influence disease outcome. Joint Bone Spine 2000, 67, 30–39. [Google Scholar] [PubMed]
- Richaud-Patin, Y.; Vega-Boada, F.; Vidaller, A.; Llorente, L. Multidrug resistance-1 (MDR-1) in autoimmune disorders IV. P-glycoprotein overfunction in lymphocytes from myasthenia gravis patients. Biomed. Pharmacother. 2004, 58, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.I.; Carlstedt-Duke, J. Involvement of multidrug resistance proteins (MDR) in the modulation of glucocorticoid response. J. Steroid Biochem. Mol. Biol. 2002, 82, 277–288. [Google Scholar]
- Hew, M.; Bhavsar, P.; Torrego, A.; Meah, S.; Khorasani, N.; Barnes, P.J.; Adcock, I.; Chung, K.F. Relative corticosteroid insensitivity of peripheral blood mononuclear cells in severe asthma. Am. J. Respir. Crit. Care Med. 2006, 174, 134–141. [Google Scholar]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar]
- Neal, G.E. The effect of aflatoxin B 1 on normal and cortisol-stimulated rat liver ribonucleic acid synthesis. Biochem. J. 1972, 130, 619–629. [Google Scholar]
- Kensler, T.W.; Busby, W.F., Jr.; Davidson, N.E.; Wogan, G.N. Aflatoxin inhibition of glucocorticoid finding capacity of rat liver nuclei. Biochim. Biophys. Acta 1976, 437, 200–210. [Google Scholar]
- Kensler, T.W.; Busby, W.F., Jr.; Davidson, N.E.; Wogan, G.N. Effect of hepatocarcinogens on the binding of glucocorticoid-receptor complex in rat liver nuclei. Cancer Res. 1976, 36, 4647–4651. [Google Scholar]
- Horikoshi, N.; Tashiro, F.; Tanaka, N.; Ueno, Y. Modulation of hormonal induction of tyrosine aminotransferase and glucocorticoid receptors by aflatoxin B1 and sterigmatocystin in Reuber hepatoma cells. Cancer Res. 1988, 48, 5188–5192. [Google Scholar]
- Wogan, G.N.; Friedman, M.A. Inhibition by aflatonin B-1 of hydrocortisone induction of rat liver tryptophan pyrrolase and tyrosine transaminase. Arch. Biochem. Biophys. 1968, 128, 509–516. [Google Scholar]
- Webster, J.I.; Tonelli, L.H.; Moayeri, M.; Simons, S.S., Jr.; Leppla, S.H.; Sternberg, E.M. Anthrax lethal factor represses glucocorticoid and progesterone receptor activity. Proc. Natl. Acad. Sci. USA 2003, 100, 5706–5711. [Google Scholar]
- Webster, J.I.; Sternberg, E.M. Anthrax lethal toxin represses glucocorticoid receptor (GR) transactivation by inhibiting GR-DNA binding in vivo. Mol. Cell. Endocrinol. 2005, 241, 21–31. [Google Scholar]
- Tait, A.S.; Dalton, M.; Geny, B.; D'Agnillo, F.; Popoff, M.R.; Sternberg, E.M. The large clostridial toxins from Clostridium sordellii and C. difficile repress glucocorticoid receptor activity. Infect. Immun. 2007, 75, 3935–3940. [Google Scholar] [PubMed]
- McCallum, R.E.; Seale, T.W.; Stith, R.D. Influence of endotoxin treatment on dexamethasone induction of hepatic phosphoenolpyruvate carboxykinase. Infect. Immun. 1983, 39, 213–219. [Google Scholar]
- Stith, R.D.; McCallum, R.E. General effect of endotoxin on glucocorticoid receptors in mammalian tissues. Circ. Shock 1986, 18, 301–309. [Google Scholar]
- Stith, R.D.; McCallum, R.E. Down regulation of hepatic glucocorticoid receptors after endotoxin treatment. Infect. Immun. 1983, 40, 613–621. [Google Scholar]
- Vaptzarova, K.I.; Baramova, E.N.; Popov, P.G. Endotoxin inhibition of glucocorticoid enzyme induction and in vivo 3H-dexamethasone labelling of rat liver nuclei. Int. J. Biochem. 1989, 21, 701–705. [Google Scholar]
- Hill, M.R.; Stith, R.D.; McCallum, R.E. Monokines mediate decreased hepatic glucocorticoid binding in endotoxemia. J. Leukoc. Biol. 1987, 41, 236–241. [Google Scholar]
- Jiayi, D.; Chen, Y.Z. LPS-induced decrease of specific binding of 3H-dexamethasone to peritoneal macrophages of C57BL/6 mice. J. Recept. Res. 1992, 12, 451–462. [Google Scholar]
- Li, F.; Xu, R.B. Changes in canine leukocyte glucocorticoid receptors during endotoxin shock. Circ. Shock 1988, 26, 99–105. [Google Scholar]
- Liu, L.Y.; Sun, B.; Tian, Y.; Lu, B.Z.; Wang, J. Changes of pulmonary glucocorticoid receptor and phospholipase A2 in sheep with acute lung injury after high dose endotoxin infusion. Am. Rev. Respir. Dis. 1993, 148, 878–881. [Google Scholar]
- Verheggen, M.M.; van Hal, P.T.; Adriaansen-Soeting, P.W.; Goense, B.J.; Hoogsteden, H.C.; Brinkmann, A.O.; Versnel, M.A. Modulation of glucocorticoid receptor expression in human bronchial epithelial cell lines by IL-1 beta, TNF-alpha and LPS. Eur. Respir. J. 1996, 9, 2036–2043. [Google Scholar]
- Basta-Kaim, A.; Budziszewska, B.; Jaworska-Feil, L.; Leskiewicz, M.; Tetich, M.; Kubera, M.; Scharpe, S.; Lason, W. Opposite effects of clozapine and sulpiride on the lipopolysaccharide-induced inhibition of the GR-mediated gene transcription in fibroblast cells. Pol. J. Pharmacol. 2003, 55, 1153–1158. [Google Scholar]
- Basta-Kaim, A.; Budziszewska, B.; Jaworska-Feil, L.; Tetich, M.; Kubera, M.; Zajicova, A.; Holan, V.; Lason, W. Effects of lipopolysaccharide and chlorpromazine on glucocorticoid receptor-mediated gene transcription and immunoreactivity: A possible involvement of p38-MAP kinase. Eur. Neuropsychopharmacol. 2004, 14, 521–528. [Google Scholar]
- Budziszewska, B.; Basta-Kaim, A.; Kubera, M.; Jaworska, L.; Leskiewicz, M.; Tetich, M.; Otczyk, M.; Zajicova, A.; Holan, V.; Lason, W. Effect of lipopolysaccharide and antidepressant drugs on glucocorticoid receptor-mediated gene transcription. Pharmacol. Rep. 2005, 57, 540–544. [Google Scholar]
- Salkowski, C.A.; Vogel, S.N. Lipopolysaccharide increases glucocorticoid receptor expression in murine macrophages. A possible mechanism for glucocorticoid-mediated suppression of endotoxicity. J. Immunol. 1992, 149, 4041–4047. [Google Scholar] [PubMed]
- Shackleford, G.M.; Hart, S.F.; Berry, L.J. Endotoxin treatment inhibits glucocorticoid induction of hepatic enzymes at a late induction step. Am. J. Physiol. 1986, 250, E218–E225. [Google Scholar]
- Hauk, P.J.; Hamid, Q.A.; Chrousos, G.P.; Leung, D.Y. Induction of corticosteroid insensitivity in human PBMCs by microbial superantigens. J. Allergy Clin. Immunol. 2000, 105, 782–787. [Google Scholar]
- Li, L.B.; Goleva, E.; Hall, C.F.; Ou, L.S.; Leung, D.Y. Superantigen-induced corticosteroid resistance of human T cells occurs through activation of the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK-ERK) pathway. J. Allergy Clin. Immunol. 2004, 114, 1059–1069. [Google Scholar]
- Fukushima, H.; Hirano, T.; Oka, K. Staphylococcus aureus-superantigen decreases FKBP51 mRNA expression and cell-response to suppressive efficacy of a glucocorticoid in human peripheral blood mononuclear cells: Possible implication of mitogen-activated protein kinase pathways. Eur. J. Pharmacol. 2007, 570, 222–228. [Google Scholar]
- Fakhri, S.; Christodoulopoulos, P.; Tulic, M.; Fukakusa, M.; Frenkiel, S.; Leung, D.Y.; Hamid, Q.A. Role of microbial toxins in the induction of glucocorticoid receptor beta expression in an explant model of rhinosinusitis. J. Otolaryngol. 2003, 32, 388–393. [Google Scholar]
- Fakhri, S.; Tulic, M.; Christodoulopoulos, P.; Fukakusa, M.; Frenkiel, S.; Leung, D.Y.; Hamid, Q.A. Microbial superantigens induce glucocorticoid receptor beta and steroid resistance in a nasal explant model. Laryngoscope 2004, 114, 887–892. [Google Scholar]
- Klimpel, K.R.; Arora, N.; Leppla, S.H. Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol. Microbiol. 1994, 13, 1093–1100. [Google Scholar]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; Vande Woude, G.F. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar]
- Pellizzari, R.; Guidi-Rontani, C.; Vitale, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999, 462, 199–204. [Google Scholar]
- Pellizzari, R.; Guidi-Rontani, C.; Vitale, G.; Mock, M.; Montecucco, C. Lethal factor of Bacillus anthracis cleaves the N-terminus of MAPKKs: Analysis of the intracellular consequences in macrophages. Int. J. Med. Microbiol. 2000, 290, 421–427. [Google Scholar]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352 Pt 3, 739–745. [Google Scholar]
- Kang, Z.; Webster Marketon, J.I.; Johnson, A.; Sternberg, E.M. Bacillus anthracis lethal toxin represses MMTV promoter activity through transcription factors. J. Mol. Biol. 2009, 389, 595–605. [Google Scholar]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar]
- Goodrum, K.J.; Berry, L.J. The use of Reuber hepatoma cells for the study of a lipopolysaccharide-induced macrophage factor: Glucocorticoid-antagonizing factor. Lab. Invest. 1979, 41, 174–181. [Google Scholar]
- Sakaguchi, S.; Ibata, H.; Yokota, K. Participation of calcium ion on depletion mechanism of liver glycogen by purified glucocorticoid antagonizing factor released in blood during endotoxemia. Microbiol. Immunol. 1990, 34, 985–994. [Google Scholar]
- Goodrum, K.J.; Berry, L.J. The effect of glucocorticoid antagonizing factor on hepatoma cells. Proc. Soc. Exp. Biol. Med. 1978, 159, 359–363. [Google Scholar]
- Sakaguchi, S.; Yokota, K. Purification and characteristics of glucocorticoid antagonizing factor in endotoxemia. Microbiol. Immunol. 1987, 31, 509–520. [Google Scholar]
- Koj, A. Initiation of acute phase response and synthesis of cytokines. Biochim. Biophys. Acta 1996, 1317, 84–94. [Google Scholar]
- Van Amersfoort, E.S.; Van Berkel, T.J.; Kuiper, J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin. Microbiol. Rev. 2003, 16, 379–414. [Google Scholar]
- Calandra, T.; Bucala, R. Macrophage migration inhibitory factor (MIF): A glucocorticoid counter-regulator within the immune system. Crit. Rev. Immunol. 1997, 17, 77–88. [Google Scholar]
- Hill, M.R.; Stith, R.D.; McCallum, R.E. Interleukin 1: A regulatory role in glucocorticoid-regulated hepatic metabolism. J. Immunol. 1986, 137, 858–862. [Google Scholar]
- Hill, M.R.; Stith, R.D.; McCallum, R.E. Human recombinant IL-1 alters glucocorticoid receptor function in Reuber hepatoma cells. J. Immunol. 1988, 141, 1522–1528. [Google Scholar]
- Kam, J.C.; Szefler, S.J.; Surs, W.; Sher, E.R.; Leung, D.Y. Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoids. J. Immunol. 1993, 151, 3460–3466. [Google Scholar]
- Spahn, J.D.; Szefler, S.J.; Surs, W.; Doherty, D.E.; Nimmagadda, S.R.; Leung, D.Y. A novel action of IL-13: induction of diminished monocyte glucocorticoid receptor-binding affinity. J. Immunol. 1996, 157, 2654–2659. [Google Scholar]
- Pariante, C.M.; Pearce, B.D.; Pisell, T.L.; Sanchez, C.I.; Po, C.; Su, C.; Miller, A.H. The proinflammatory cytokine, interleukin-1alpha, reduces glucocorticoid receptor translocation and function. Endocrinology 1999, 140, 4359–4366. [Google Scholar]
- Rakasz, E.; Gal, A.; Biro, J.; Balas, G.; Falus, A. Modulation of glucocorticosteroid binding in human lymphoid, monocytoid and hepatoma cell lines by inflammatory cytokines interleukin (IL)-1 beta, IL-6 and tumour necrosis factor (TNF)-alpha. Scand. J. Immunol. 1993, 37, 684–689. [Google Scholar]
- Salkowski, C.A.; Vogel, S.N. IFN-gamma mediates increased glucocorticoid receptor expression in murine macrophages. J. Immunol. 1992, 148, 2770–2777. [Google Scholar]
- Jonat, C.; Rahmsdorf, H.J.; Park, K.K.; Cato, A.C.; Gebel, S.; Ponta, H.; Herrlich, P. Antitumor promotion and antiinflammation: Down-modulation of AP-1 (Fos/Jun) activity by glucocorticoid hormone. Cell 1990, 62, 1189–1204. [Google Scholar]
- Schule, R.; Rangarajan, P.; Kliewer, S.; Ransone, L.J.; Bolado, J.; Yang, N.; Verma, I.M.; Evans, R.M. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell 1990, 62, 1217–1226. [Google Scholar]
- Yang-Yen, H.F.; Chambard, J.C.; Sun, Y.L.; Smeal, T.; Schmidt, T.J.; Drouin, J.; Karin, M. Transcriptional interference between c-Jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell 1990, 62, 1205–1215. [Google Scholar]
- McKay, L.I.; Cidlowski, J.A. Cross-talk between nuclear factor-kappa B and the steroid hormone receptors: Mechanisms of mutual antagonism. Mol. Endocrinol. 1998, 12, 45–56. [Google Scholar]
- Clifford, J.I.; Rees, K.R. The action of aflatoxin B1 on the rat liver. Biochem. J. 1967, 102, 65–75. [Google Scholar]
- van Aswegen, C.H.; Lewko, W.M.; Wittliff, J.L. Influence of phomopsin and ivalin on steroid-hormone binding and growth of MCF-7 human breast cancer cells. J. Toxicol. Environ. Health 1985, 16, 13–23. [Google Scholar]
- van Aswegen, C.H.; Wittliff, J.L. Steroid hormone-receptor activity in the presence of a mycotoxic phomopsin toxin and the sesquiterpene ivalin toxin. J. Toxicol. Environ. Health 1985, 16, 1–12. [Google Scholar]
- Barr, F.D.; Krohmer, L.J.; Hamilton, J.W.; Sheldon, L.A. Disruption of histone modification and CARM1 recruitment by arsenic represses transcription at glucocorticoid receptor-regulated promoters. PLoS One 2009, 4, e6766. [Google Scholar]
- Bodwell, J.E.; Gosse, J.A.; Nomikos, A.P.; Hamilton, J.W. Arsenic disruption of steroid receptor gene activation: Complex dose-response effects are shared by several steroid receptors. Chem. Res. Toxicol. 2006, 19, 1619–1629. [Google Scholar]
- Bodwell, J.E.; Kingsley, L.A.; Hamilton, J.W. Arsenic at very low concentrations alters glucocorticoid receptor (GR)-mediated gene activation but not GR-mediated gene repression: complex dose-response effects are closely correlated with levels of activated GR and require a functional GR DNA binding domain. Chem. Res. Toxicol. 2004, 17, 1064–1076. [Google Scholar]
- Hamilton, J.W.; Kaltreider, R.C.; Bajenova, O.V.; Ihnat, M.A.; McCaffrey, J.; Turpie, B.W.; Rowell, E.E.; Oh, J.; Nemeth, M.J.; Pesce, C.A.; Lariviere, J.P. Molecular basis for effects of carcinogenic heavy metals on inducible gene expression. Environ. Health Perspect. 1998, 106 Suppl 4, 1005–1015. [Google Scholar]
- Kaltreider, R.C.; Davis, A.M.; Lariviere, J.P.; Hamilton, J.W. Arsenic alters the function of the glucocorticoid receptor as a transcription factor. Environ. Health Perspect. 2001, 109, 245–251. [Google Scholar]
- Dundjerski, J.; Butorovic, B.; Kipic, J.; Trajkovic, D.; Matic, G. Cadmium affects the activity of rat liver tyrosine aminotransferase and its induction by dexamethasone. Arch. Toxicol. 1996, 70, 390–395. [Google Scholar]
- Dundjerski, J.; Kovac, T.; Pavkovic, N.; Cvoro, A.; Matic, G. Glucocorticoid receptor-Hsp90 interaction in the liver cytosol of cadmium-intoxicated rats. Cell. Biol. Toxicol. 2000, 16, 375–383. [Google Scholar]
- Lopez, S.; Miyashita, Y.; Simons, S.S., Jr. Structurally based, selective interaction of arsenite with steroid receptors. J. Biol. Chem. 1990, 265, 16039–16042. [Google Scholar]
- Simons, S.S., Jr.; Chakraborti, P.K.; Cavanaugh, A.H. Arsenite and cadmium(II) as probes of glucocorticoid receptor structure and function. J. Biol. Chem. 1990, 265, 1938–1945. [Google Scholar]
- Stancato, L.F.; Hutchison, K.A.; Chakraborti, P.K.; Simons, S.S., Jr.; Pratt, W.B. Differential effects of the reversible thiol-reactive agents arsenite and methyl methanethiosulfonate on steroid binding by the glucocorticoid receptor. Biochemistry 1993, 32, 3729–3736. [Google Scholar]
- Perry, S.T.; Kulkarni, S.B.; Lee, K.L.; Kenney, F.T. Selective effect of the metallocarcinogen beryllium on hormonal regulation of gene expression in cultured cells. Cancer Res. 1982, 42, 473–476. [Google Scholar]
- Ord, M.G.; Stocken, L.A. Enzyme induction in rat liver: the effects of Be2+ in vivo. Biosci. Rep. 1981, 1, 217–222. [Google Scholar]
- Dunderski, J.; Stanosevic, J.; Ristic, B.; Trajkovic, D.; Matic, G. In vivo effects of cadmium on rat liver glucocorticoid receptor functional properties. Int. J. Biochem. 1992, 24, 1065–1072. [Google Scholar]
- McCaffrey, J.; Wolf, C.M.; Hamilton, J.W. Effects of the genotoxic carcinogen chromium(VI) on basal and hormone-inducible phosphoenolpyruvate carboxykinase gene expression in vivo: Correlation with glucocorticoid- and developmentally regulated expression. Mol. Carcinog. 1994, 10, 189–198. [Google Scholar]
- Heiman, A.S.; Tonner, L.E. The acute effect of lead acetate on glucocorticoid regulation of tyrosine aminotransferase in hepatoma cells. Toxicology 1995, 100, 57–68. [Google Scholar]
- Dundjerski, J.; Brkljacic, J.; Elakovic, I.; Manitasevic, S.; Matic, G. Mercury influences rat liver tyrosine aminotransferase activity and induction by dexamethasone. J. Appl. Toxicol. 2006, 26, 187–190. [Google Scholar]
- Brkljacic, J.; Milutinovic, D.V.; Dundjerski, J.; Matic, G. Mercury inhibits rat liver and kidney glucocorticoid receptor hormone binding activity. Cell. Biol. Toxicol. 2004, 20, 171–182. [Google Scholar]
- Brkljacic, J.; Milutinovic, D.V.; Dundjerski, J.; Matic, G. Mercury stimulates rat liver glucocorticoid receptor association with Hsp90 and Hsp70. J. Biochem. Mol. Toxicol. 2004, 18, 257–260. [Google Scholar]
- DeMoor, J.M.; Kennette, W.A.; Collins, O.M.; Koropatnick, J. Zinc-metallothionein levels are correlated with enhanced glucocorticoid responsiveness in mouse cells exposed to ZnCl(2), HgCl(2), and heat shock. Toxicol. Sci. 2001, 64, 67–76. [Google Scholar]
- Tashima, Y.; Terui, M.; Itoh, H.; Mizunuma, H.; Kobayashi, R.; Marumo, F. Effect of selenite on glucocorticoid receptor. J. Biochem. 1989, 105, 358–361. [Google Scholar]
- Telford, W.G.; Fraker, P.J. Zinc reversibly inhibits steroid binding to murine glucocorticoid receptor. Biochem. Biophys. Res. Commun. 1997, 238, 86–89. [Google Scholar]
- Verheggen, M.M.; Adriaansen-Soeting, P.W.; Berrevoets, C.A.; van Hal, P.T.; Brinkmann, A.O.; Hoogsteden, H.C.; Versnel, M.A. Glucocorticoid receptor expression in human bronchial epithelial cells: effects of smoking and COPD. Mediators Inflamm. 1998, 7, 275–281. [Google Scholar]
- Renkema, T.E.; Schouten, J.P.; Koeter, G.H.; Postma, D.S. Effects of long-term treatment with corticosteroids in COPD. Chest 1996, 109, 1156–1162. [Google Scholar]
- Marwick, J.A.; Caramori, G.; Stevenson, C.S.; Casolari, P.; Jazrawi, E.; Barnes, P.J.; Ito, K.; Adcock, I.M.; Kirkham, P.A.; Papi, A. Inhibition of PI3Kdelta restores glucocorticoid function in smoking-induced airway inflammation in mice. Am. J. Respir. Crit. Care Med. 2009, 179, 542–548. [Google Scholar]
- Livingston, E.; Darroch, C.E.; Chaudhuri, R.; McPhee, I.; McMahon, A.D.; Mackenzie, S.J.; Thomson, N.C. Glucocorticoid receptor alpha:beta ratio in blood mononuclear cells is reduced in cigarette smokers. J. Allergy Clin. Immunol. 2004, 114, 1475–1478. [Google Scholar]
- Hukkanen, J.; Vaisanen, T.; Lassila, A.; Piipari, R.; Anttila, S.; Pelkonen, O.; Raunio, H.; Hakkola, J. Regulation of CYP3A5 by glucocorticoids and cigarette smoke in human lung-derived cells. J. Pharmacol. Exp. Ther. 2003, 304, 745–752. [Google Scholar]
- Xu, H.; Ferro, T.J.; Chu, S. Cigarette smoke condensate inhibits ENaC alpha-subunit expression in lung epithelial cells. Eur. Respir. J. 2007, 30, 633–642. [Google Scholar]
- Ito, K.; Lim, S.; Caramori, G.; Chung, K.F.; Barnes, P.J.; Adcock, I.M. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophages. FASEB J. 2001, 15, 1110–1112. [Google Scholar]
- Alam, M.G.; Allinson, G.; Stagnitti, F.; Tanaka, A.; Westbrooke, M. Arsenic contamination in Bangladesh groundwater: A major environmental and social disaster. Int. J. Environ. Health Res. 2002, 12, 235–253. [Google Scholar]
- Phillips, K.P.; Foster, W.G. Key developments in endocrine disrupter research and human health. J. Toxicol. Environ Health B. Crit. Rev. 2008, 11, 322–344. [Google Scholar]
- Abe, J.; Kotzin, B.L.; Meissner, C.; Melish, M.E.; Takahashi, M.; Fulton, D.; Romagne, F.; Malissen, B.; Leung, D.Y. Characterization of T cell repertoire changes in acute Kawasaki disease. J. Exp. Med. 1993, 177, 791–796. [Google Scholar]
- Athappan, G.; Gale, S.; Ponniah, T. Corticosteroid therapy for primary treatment of Kawasaki disease - weight of evidence: A meta-analysis and systematic review of the literature. Cardiovasc. J. Afr. 2009, 20, 233–236. [Google Scholar]
- Paliard, X.; West, S.G.; Lafferty, J.A.; Clements, J.R.; Kappler, J.W.; Marrack, P.; Kotzin, B.L. Evidence for the effects of a superantigen in rheumatoid arthritis. Science 1991, 253, 325–329. [Google Scholar]
- Hauk, P.J.; Wenzel, S.E.; Trumble, A.E.; Szefler, S.J.; Leung, D.Y. Increased T-cell receptor vbeta8+ T cells in bronchoalveolar lavage fluid of subjects with poorly controlled asthma: a potential role for microbial superantigens. J. Allergy Clin. Immunol. 1999, 104, 37–45. [Google Scholar]
- Strickland, I.; Hauk, P.J.; Trumble, A.E.; Picker, L.J.; Leung, D.Y. Evidence for superantigen involvement in skin homing of T cells in atopic dermatitis. J. Invest. Dermatol. 1999, 112, 249–253. [Google Scholar]
- Barnes, P.J.; Adcock, I.M. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917. [Google Scholar]
- Hagg, P.M.; Hurskainen, T.; Palatsi, R.; Ilves, M.; Oikarinen, A. Increased expression of glucocorticoid receptor beta in lymphocytes of patients with severe atopic dermatitis unresponsive to topical corticosteroid. Br. J. Dermatol. 2010, 162, 318–324. [Google Scholar]
- Loke, T.K.; Sousa, A.R.; Corrigan, C.J.; Lee, T.H. Glucocorticoid-resistant asthma. Curr. Allergy Asthma Rep. 2002, 2, 144–150. [Google Scholar]
- Silverman, M.N.; Sternberg, E.M. Neuroendocrine-immune interactions in rheumatoid arthritis: mechanisms of glucocorticoid resistance. Neuroimmunomodulation 2008, 15, 19–28. [Google Scholar]
- Cohen, A.L.; Bhatnagar, J.; Reagan, S.; Zane, S.B.; D'Angeli, M.A.; Fischer, M.; Killgore, G.; Kwan-Gett, T.S.; Blossom, D.B.; Shieh, W.J.; Guarner, J.; Jernigan, J.; Duchin, J.S.; Zaki, S.R.; McDonald, L.C. Toxic shock associated with Clostridium sordellii and Clostridium perfringens after medical and spontaneous abortion. Obstet. Gynecol. 2007, 110, 1027–1033. [Google Scholar]
- Fischer, M.; Bhatnagar, J.; Guarner, J.; Reagan, S.; Hacker, J.K.; Van Meter, S.H.; Poukens, V.; Whiteman, D.B.; Iton, A.; Cheung, M.; Dassey, D.E.; Shieh, W.J.; Zaki, S.R. Fatal toxic shock syndrome associated with Clostridium sordellii after medical abortion. N. Engl. J. Med. 2005, 353, 2352–2360. [Google Scholar]
- McGregor, J.A.; Soper, D.E.; Lovell, G.; Todd, J.K. Maternal deaths associated with Clostridium sordellii infection. Am. J. Obstet. Gynecol. 1989, 161, 987–995. [Google Scholar]
- Miech, R.P. Pathophysiology of mifepristone-induced septic shock due to Clostridium sordellii. Ann Pharmacother 2005, 39, 1483–1488. [Google Scholar]
- Sinave, C.; Le Templier, G.; Blouin, D.; Leveille, F.; Deland, E. Toxic shock syndrome due to Clostridium sordellii: A dramatic postpartum and postabortion disease. Clin. Infect. Dis. 2002, 35, 1441–1443. [Google Scholar]
- Barnes, P.J. Chronic obstructive pulmonary disease: new opportunities for drug development. Trends Pharmacol. Sci. 1998, 19, 415–423. [Google Scholar]
- Van Overveld, F.J.; Demkow, U.; Gorecka, D.; De Backer, W.A.; Zielinski, J. Differences in responses upon corticosteroid therapy between smoking and non-smoking patients with COPD. J. Physiol. Pharmacol. 2006, 57, 273–282. [Google Scholar]
- Pedersen, B.; Dahl, R.; Karlstrom, R.; Peterson, C.G.; Venge, P. Eosinophil and neutrophil activity in asthma in a one-year trial with inhaled budesonide. The impact of smoking. Am. J. Respir. Crit. Care Med. 1996, 153, 1519–1529. [Google Scholar] [PubMed]
- Chalmers, G.W.; Macleod, K.J.; Little, S.A.; Thomson, L.J.; McSharry, C.P.; Thomson, N.C. Influence of cigarette smoking on inhaled corticosteroid treatment in mild asthma. Thorax 2002, 57, 226–230. [Google Scholar]
- Chaudhuri, R.; Livingston, E.; McMahon, A.D.; Thomson, L.; Borland, W.; Thomson, N.C. Cigarette smoking impairs the therapeutic response to oral corticosteroids in chronic asthma. Am. J. Respir. Crit. Care Med. 2003, 168, 1308–1311. [Google Scholar]
- Tomlinson, J.E.; McMahon, A.D.; Chaudhuri, R.; Thompson, J.M.; Wood, S.F.; Thomson, N.C. Efficacy of low and high dose inhaled corticosteroid in smokers versus non-smokers with mild asthma. Thorax 2005, 60, 282–287. [Google Scholar]
- Livingston, E.; Chaudhuri, R.; McMahon, A.D.; Fraser, I.; McSharry, C.P.; Thomson, N.C. Systemic sensitivity to corticosteroids in smokers with asthma. Eur. Respir. J. 2007, 29, 64–71. [Google Scholar]
- Cox, G.; Whitehead, L.; Dolovich, M.; Jordana, M.; Gauldie, J.; Newhouse, M.T. A randomized controlled trial on the effect of inhaled corticosteroids on airways inflammation in adult cigarette smokers. Chest 1999, 115, 1271–1277. [Google Scholar]
- Sprung, C.L.; Goodman, S.; Weiss, Y.G. Steroid therapy of septic shock. Crit. Care Clin. 2009, 25, 825–834. [Google Scholar]
- Annane, D.; Bellissant, E.; Bollaert, P.E.; Briegel, J.; Confalonieri, M.; De Gaudio, R.; Keh, D.; Kupfer, Y.; Oppert, M.; Meduri, G.U. Corticosteroids in the treatment of severe sepsis and septic shock in adults: A systematic review. JAMA 2009, 301, 2362–2375. [Google Scholar]
- Annane, D.; Maxime, V.; Ibrahim, F.; Alvarez, J.C.; Abe, E.; Boudou, P. Diagnosis of adrenal insufficiency in severe sepsis and septic shock. Am. J. Respir. Crit. Care Med. 2006, 174, 1319–1326. [Google Scholar]
- Maxime, V.; Lesur, O.; Annane, D. Adrenal insufficiency in septic shock. Clin. Chest Med. 2009, 30, 17–27. [Google Scholar]
- Prigent, H.; Maxime, V.; Annane, D. Science review: mechanisms of impaired adrenal function in sepsis and molecular actions of glucocorticoids. Crit. Care 2004, 8, 243–252. [Google Scholar]
- Molijn, G.J.; Spek, J.J.; van Uffelen, J.C.; de Jong, F.H.; Brinkmann, A.O.; Bruining, H.A.; Lamberts, S.W.; Koper, J.W. Differential adaptation of glucocorticoid sensitivity of peripheral blood mononuclear leukocytes in patients with sepsis or septic shock. J. Clin. Endocrinol. Metab. 1995, 80, 1799–1803. [Google Scholar]
- Molijn, G.J.; Koper, J.W.; van Uffelen, C.J.; de Jong, F.H.; Brinkmann, A.O.; Bruining, H.A.; Lamberts, S.W. Temperature-induced down-regulation of the glucocorticoid receptor in peripheral blood mononuclear leucocyte in patients with sepsis or septic shock. Clin. Endocrinol. 1995, 43, 197–203. [Google Scholar]
- Mastorci, F.; Vicentini, M.; Viltart, O.; Manghi, M.; Graiani, G.; Quaini, F.; Meerlo, P.; Nalivaiko, E.; Maccari, S.; Sgoifo, A. Long-term effects of prenatal stress: changes in adult cardiovascular regulation and sensitivity to stress. Neurosci. Biobehav. Rev. 2009, 33, 191–203. [Google Scholar]
- Haczku, A.; Panettieri, R.A., Jr. Social stress and asthma: The role of corticosteroid insensitivity. J. Allergy Clin. Immunol. 2010, 125, 550–558. [Google Scholar]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Webster Marketon, J.I.; Sternberg, E.M. The Glucocorticoid Receptor: A Revisited Target for Toxins. Toxins 2010, 2, 1357-1380. https://doi.org/10.3390/toxins2061357
Webster Marketon JI, Sternberg EM. The Glucocorticoid Receptor: A Revisited Target for Toxins. Toxins. 2010; 2(6):1357-1380. https://doi.org/10.3390/toxins2061357
Chicago/Turabian StyleWebster Marketon, Jeanette I., and Esther M. Sternberg. 2010. "The Glucocorticoid Receptor: A Revisited Target for Toxins" Toxins 2, no. 6: 1357-1380. https://doi.org/10.3390/toxins2061357