Patulin Alters Insulin Signaling and Metabolic Flexibility in HepG2 and HEK293 Cells


Abstract
:1. Introduction
2. Results
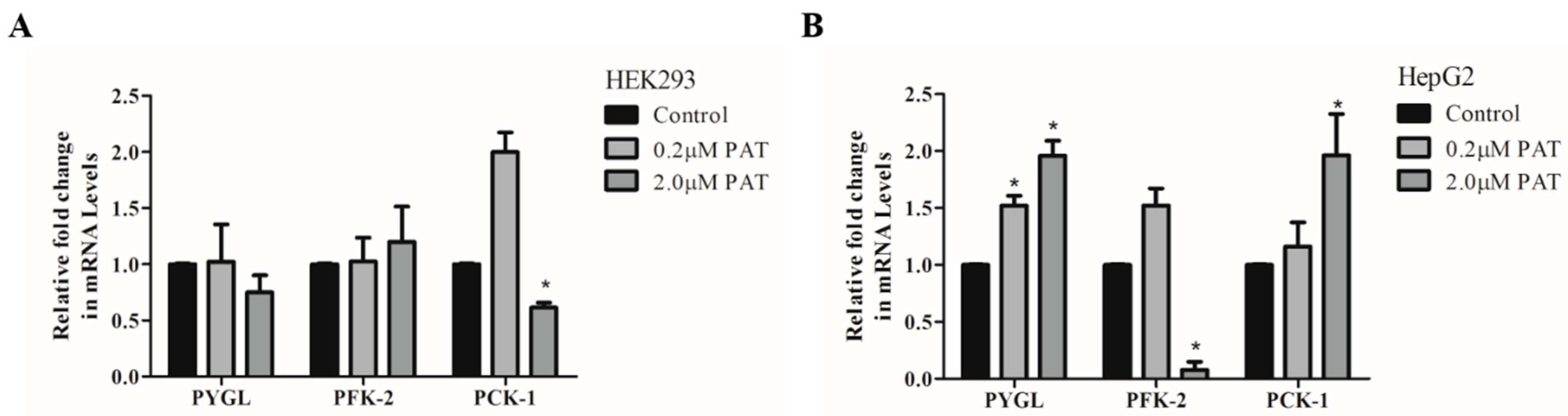
2.1. PAT Alters Expression of Enzymes Involved in Glucose Generation
2.2. Glucose Availability Determines PAT-Associated Insulin Signaling Defects in HepG2 Cells
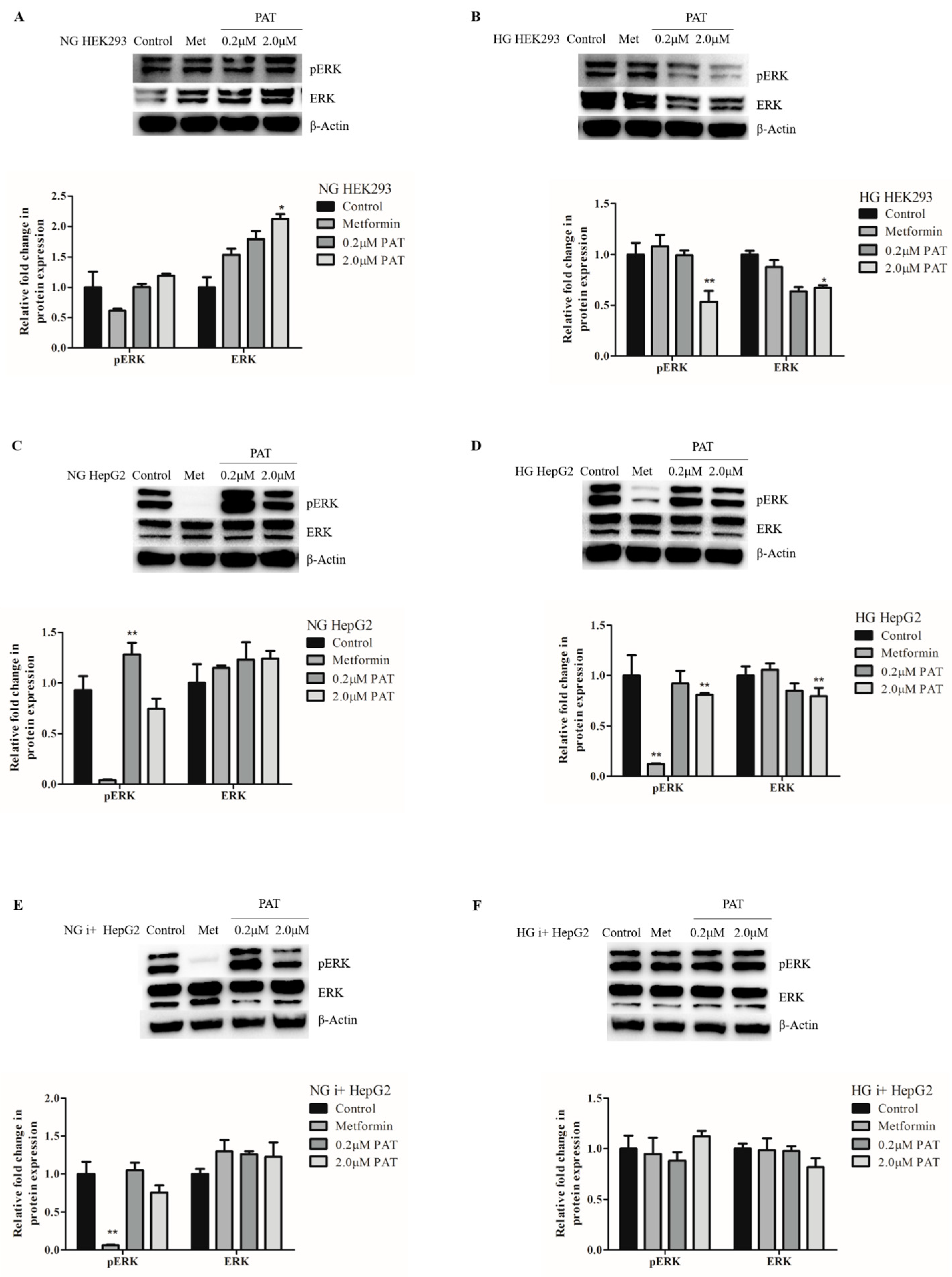
2.3. PAT-Altered ERK/MAPK Signaling Is Influenced by Glucose and Insulin Availability
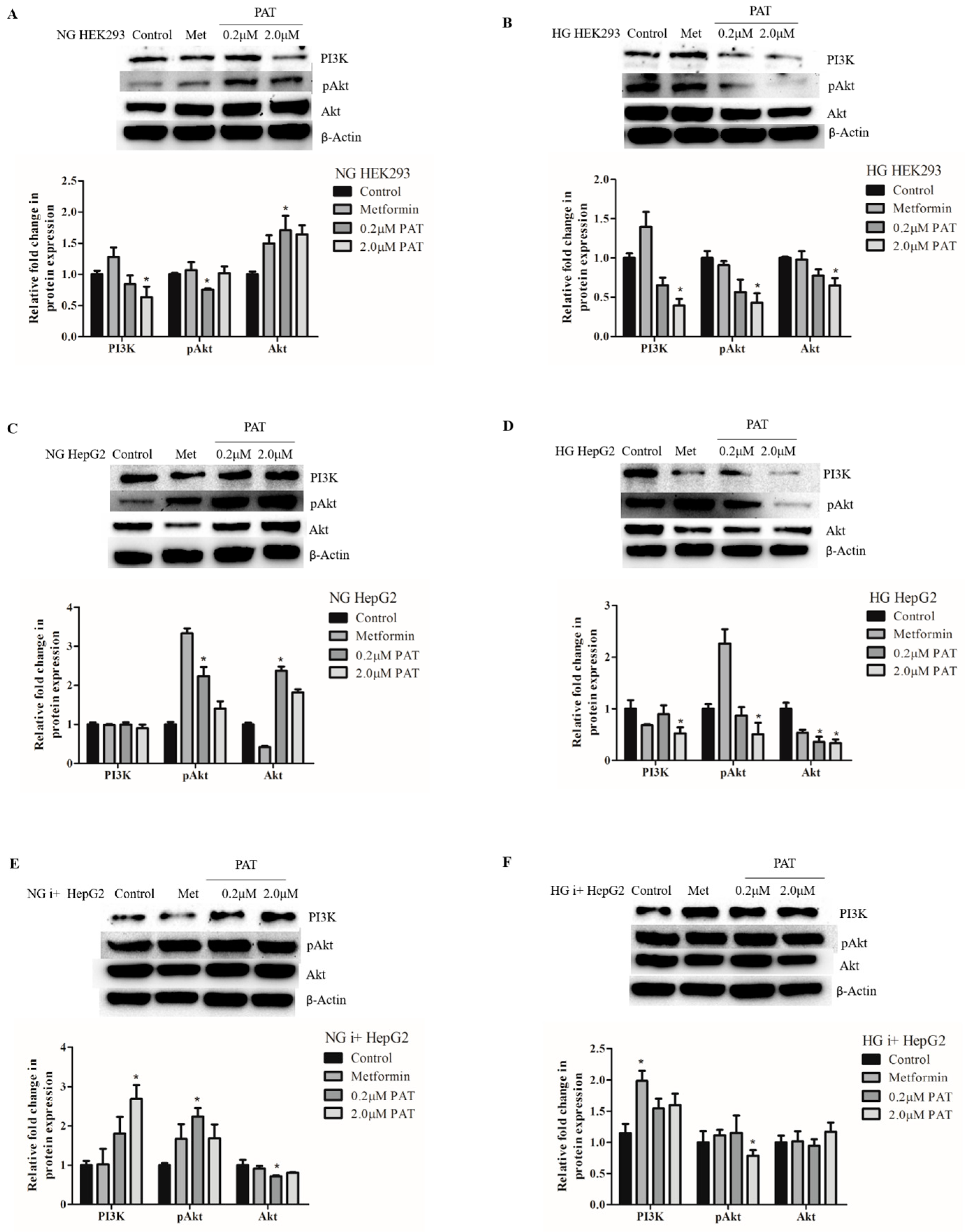
2.4. Glucose Availability Affects PAT-Mediated PI3K/Akt Signaling Trends
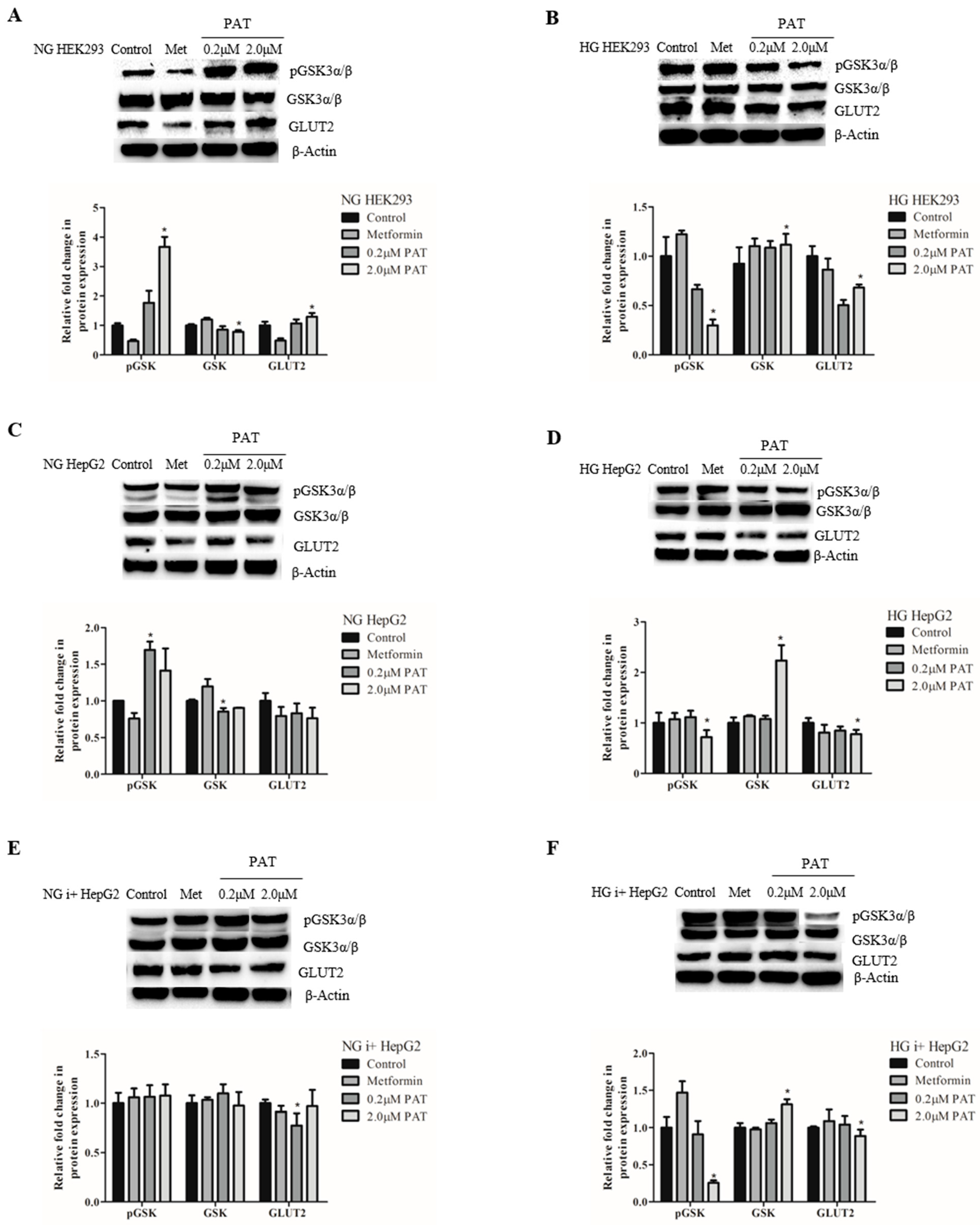
2.5. PAT-Modified GSK-3 Activation Corresponded with PI3K/Akt Signaling Trends While GLUT2 Expression Was Widely Compromised by PAT
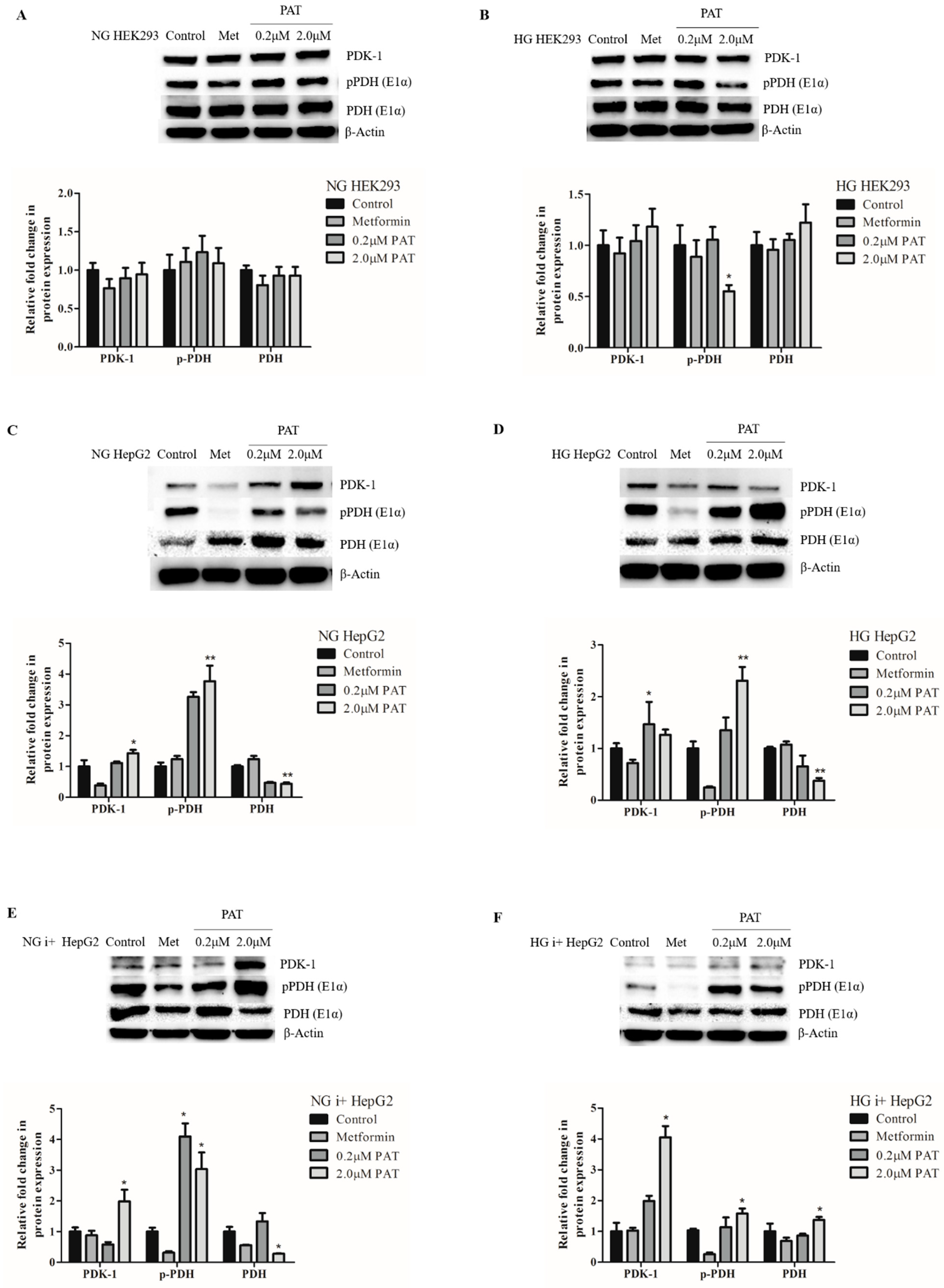
2.6. PAT Contributes to Metabolic Inflexibility by PDK-1 Elevation and PDH Inhibition under NG and HG Conditions
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture
5.2. Dosage Information
5.3. Quantitative Polymerase Chain Reaction (PCR)
5.3.1. RNA Isolation and cDNA Preparation
5.3.2. mRNA Quantification
5.4. Western Blotting
5.4.1. Protein Isolation
5.4.2. Sodium Dodecyl Sulphate–Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Immunoblotting
5.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Islam, S.M.S.; Purnat, T.D.; Phuong, N.T.A.; Mwingira, U.; Schacht, K.; Fröschl, G. Non-Communicable Diseases (NCDs) in developing countries: A symposium report. Glob. Health 2014, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.J. Overnutrition Determines LPS Regulation of Mycotoxin Induced Neurotoxicity in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 29554–29573. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Singh, N.; Ansari, K.M. Toxicological effects of patulin mycotoxin on the mammalian system: An overview. Toxicol. Res. 2017, 6, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.-H.; Wu, T.-S.; Yu, F.-Y.; Su, C.-C. Induction of Oxidative Stress Response by the Mycotoxin Patulin in Mammalian Cells. Toxicol. Sci. 2007, 95, 340–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sant’Ana, A.d.S.; Rosenthal, A.; de Massaguer, P.R. The fate of patulin in apple juice processing: A review. Food Res. Int. 2008, 41, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, E.; Gross, K.; Metzler, M. Aneuploidogenic and clastogenic potential of the mycotoxins citrinin and patulin. Carcinogenesis 1998, 19, 1313–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, I.; Oliveira, N.; Laires, A.; Rodrigues, A.; Rueff, J. Induction of micronuclei and chromosomal aberrations by the mycotoxin patulin in mammalian cells: Role of ascorbic acid as a modulator of patulin clastogenicity. Mutagenesis 2000, 15, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, D.M.; Metzler, M.; Lehmann, L. Mutagenicity of the mycotoxin patulin in cultured Chinese hamster V79 cells, and its modulation by intracellular glutathione. Arch. Toxicol. 2005, 79, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Glaser, N.; Stopper, H. Patulin: Mechanism of genotoxicity. Food Chem. Toxicol. 2012, 50, 1796–1801. [Google Scholar] [CrossRef]
- Zhang, B.; Peng, X.; Li, G.; Xu, Y.; Xia, X.; Wang, Q. Oxidative stress is involved in Patulin induced apoptosis in HEK293 cells. Toxicon 2015, 94 (Suppl. C), 1–7. [Google Scholar] [CrossRef]
- Zhong, Y.; Jin, C.; Gan, J.; Wang, X.; Shi, Z.; Xia, X.; Peng, X. Apigenin attenuates patulin-induced apoptosis in HEK293 cells by modulating ROS-mediated mitochondrial dysfunction and caspase signal pathway. Toxicon 2017, 137, 106–113. [Google Scholar] [CrossRef]
- Tsai, W.T.; Lo, Y.C.; Wu, M.S.; Li, C.Y.; Kuo, Y.P.; Lai, Y.H.; Tsai, Y.; Chen, K.C.; Chuang, T.H.; Yao, C.H.; et al. Mycotoxin patulin suppresses innate immune responses by mitochondrial dysfunction and p62/sequestosome-1-dependent mitophagy. J. Biol. Chem. 2016, 291, 19299–19311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puel, O.; Galtier, P.; Oswald, I. Biosynthesis and toxicological effects of patulin. Toxins 2010, 2, 613–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Sakthisekaran, D.; Shanmugasundaram, K.R.; Shanmugasundaram, E.R. Effect of patulin on some enzymes of carbohydrate metabolism studied in rats. Biochem. Int. 1989, 19, 37–51. [Google Scholar] [PubMed]
- Tao, R.; Xiong, X.; Harris, R.A.; White, M.F.; Dong, X.C. Genetic inactivation of pyruvate dehydrogenase kinases improves hepatic insulin resistance induced diabetes. PLoS ONE 2013, 8, e71997. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.; Kitsberg, D.; Tsytkin, S.; Shulman, M.; Aroeti, B.; Nahmias, Y. Live imaging of GLUT2 glucose-dependent trafficking and its inhibition in polarized epithelial cysts. Open Biol. 2014, 4, 140091. [Google Scholar] [CrossRef] [Green Version]
- Haeusler, R.A.; Camastra, S.; Astiarraga, B.; Nannipieri, M.; Anselmino, M.; Ferrannini, E. Decreased expression of hepatic glucokinase in type 2 diabetes. Mol. Metab. 2015, 4, 222–226. [Google Scholar] [CrossRef]
- Henriksen, E.J.; Dokken, B.B. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets 2006, 7, 1435–1441. [Google Scholar] [CrossRef]
- Maffucci, T.; Brancaccio, A.; Piccolo, E.; Stein, R.C.; Falasca, M. Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J. 2003, 22, 4178–4189. [Google Scholar] [CrossRef] [Green Version]
- Fröjdö, S.; Vidal, H.; Pirola, L. Alterations of insulin signaling in type 2 diabetes: A review of the current evidence from humans. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2009, 1792, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, S.; Pal, A.; Kumar, R.; Dwivedi, P.D.; Das, M.; Ansari, K.M. EGFR-mediated Akt and MAPKs signal pathways play a crucial role in patulin-induced cell proliferation in primary murine keratinocytes via modulation of Cyclin D1 and COX-2 expression. Mol. Carcinog. 2014, 53, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Nisr, R.B.; Russell, M.A.; Chrachri, A.; Moody, A.J.; Gilpin, M.L. Effects of the microbial secondary metabolites pyrrolnitrin, phenazine and patulin on INS-1 rat pancreatic β-cells. FEMS Immunol. Med. Microbiol. 2011, 63, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Al Seeni, M.N.; Ali, A.M.; Elsawi, N.M.; Abdo, A.S. Assessment of the Secondary Metabolite Patulin and Lycium Barbarum Fruit on INS-1 Rat Pancreatic Β-Cells. Agric. Food Sci. Res. 2017, 4, 24–29. [Google Scholar]
- Zhou, S.-M.; Jiang, L.-P.; Geng, C.-Y.; Cao, J.; Zhong, L.-F. Patulin-induced oxidative DNA damage and p53 modulation in HepG2 cells. Toxicon 2010, 55, 390–395. [Google Scholar] [CrossRef]
- Boussabbeh, M.; Ben Salem, I.; Prola, A.; Guilbert, A.; Bacha, H.; Abid-Essefi, S.; Lemaire, C. Patulin induces apoptosis through ROS-mediated endoplasmic reticulum stress pathway. Toxicol. Sci. 2015, 144, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [Green Version]
- Brozinick, J.T.; Roberts, B.R.; Dohm, G.L. Defective Signaling Through Akt-2 and -3 But Not Akt-1 in Insulin-Resistant Human Skeletal Muscle. Diabetes 2003, 52, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Devaraj, H.; Shanmugasundaram, K.; Shanmugasundaram, E. Changes in carbohydrate metabolism during Patulin toxicosis studied in chicks. J. Indian Inst. Sci. 2013, 64, 65. [Google Scholar]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhang, S.L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target? Int. J. Biol. Sci. 2015, 11, 1390–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, M.K.; Suk, K. Pyruvate Dehydrogenase Kinase as a Potential Therapeutic Target for Malignant Gliomas. Brain Tumor Res. Treat. 2013, 1, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golias, T.; Papandreou, I.; Sun, R.; Kumar, B.; Brown, N.V.; Swanson, B.J.; Pai, R.; Jaitin, D.; Le, Q.-T.; Teknos, T.N.; et al. Hypoxic repression of pyruvate dehydrogenase activity is necessary for metabolic reprogramming and growth of model tumours. Sci. Rep. 2016, 6, 31146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chain, E.; Florey, H.W.; Jennings, M.A. An antibacterial substance produced by Penicillium claviforme. Br. J. Exp. Pathol. 1942, 23, 202. [Google Scholar]
- Liste-Calleja, L.; Lecina, M.; Lopez-Repullo, J.; Albiol, J.; Solà, C.; Cairó, J.J. Lactate and glucose concomitant consumption as a self-regulated pH detoxification mechanism in HEK293 cell cultures. Appl. Microbiol. Biotechnol. 2015, 99, 9951–9960. [Google Scholar] [CrossRef]
- Mondon, C.E.; Jones, I.R.; Azhar, S.; Hollenbeck, C.B.; Reaven, G.M. Lactate production and pyruvate dehydrogenase activity in fat and skeletal muscle from diabetic rats. Diabetes 1992, 41, 1547–1554. [Google Scholar] [CrossRef]
- Montes, M.; Chicco, A.; Lombardo, Y.B. The effect of insulin on the uptake and metabolic fate of glucose in isolated perfused hearts of dyslipemic rats. J. Nutr. Biochem. 2000, 11, 30–37. [Google Scholar] [CrossRef]
- Alves, T.C.; Befroy, D.E.; Kibbey, R.G.; Kahn, M.; Codella, R.; Carvalho, R.A.; Petersen, K.F.; Shulman, G.I. Regulation of hepatic fat and glucose oxidation in rats with lipid-induced hepatic insulin resistance. Hepatology 2011, 53, 1175–1181. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Ahn, S.Y.; Song, I.-C.; Chung, M.H.; Jang, H.C.; Park, K.S.; Lee, K.-U.; Pak, Y.K.; Lee, H.K. Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PLoS ONE 2009, 4, e5186. [Google Scholar] [CrossRef]
- Gerets, H.H.J.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, Z.; Feng, J.H.; Chen, Y.B.; Liao, N.S.; Su, Y.; Zou, C.Y. Metabolic profiling of normal hepatocyte and hepatocellular carcinoma cells via (1) H nuclear magnetic resonance spectroscopy. Cell Biol. Int. 2018, 42, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Monge, I.; Albiol, J.; Lecina, M.; Liste-Calleja, L.; Miret, J.; Solà, C.; Cairó, J.J. Metabolic flux balance analysis during lactate and glucose concomitant consumption in HEK293 cell cultures. Biotechnol. Bioeng. 2019, 116, 388–404. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Lactate Levels (mmol/L) Normoglycemic Media (NG) | Lactate Levels (mmol/L) Hyperglycemic Media (HG) |
|---|---|---|
| HEK293 Control | 8.85 ± 0.1306 | 17.78 ± 0.07348 |
| HEK293 0.2 μM PAT | 7.77 ± 0.1796 | 12.92 ± 0.1878 |
| HEK293 2.0 μM PAT | 7.75 ± 0.3103 | 15.97 ± 0.1470 |
| HepG2 Control | 3.36 ± 0.2531 | 5.58 ± 0.2368 |
| HepG2 0.2 µM PAT | 2.64 ± 0.1551 | 5.35 ± 0.3101 |
| HepG2 2.0 µM PAT | 3.12 ± 0.1796 | 5.17 ± 0.8410 |
| HepG2 (i+) Control | 3.15 ± 0.1388 | 3.63 ± 0.07348 |
| HepG2 (i+) 0.2 µM PAT | 2.79 ± 0.1470 | 3.69 ± 0.4736 |
| HepG2 (i+) 2.0 µM PAT | 2.40 ± 0.04899 | 3.68 ± 0.2286 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pillay, Y.; Nagiah, S.; Chuturgoon, A. Patulin Alters Insulin Signaling and Metabolic Flexibility in HepG2 and HEK293 Cells. Toxins 2023, 15, 244. https://doi.org/10.3390/toxins15040244
Pillay Y, Nagiah S, Chuturgoon A. Patulin Alters Insulin Signaling and Metabolic Flexibility in HepG2 and HEK293 Cells. Toxins. 2023; 15(4):244. https://doi.org/10.3390/toxins15040244
Chicago/Turabian StylePillay, Yashodani, Savania Nagiah, and Anil Chuturgoon. 2023. "Patulin Alters Insulin Signaling and Metabolic Flexibility in HepG2 and HEK293 Cells" Toxins 15, no. 4: 244. https://doi.org/10.3390/toxins15040244