Uremic Toxins and Cardiovascular Risk in Chronic Kidney Disease: What Have We Learned Recently beyond the Past Findings?



Abstract
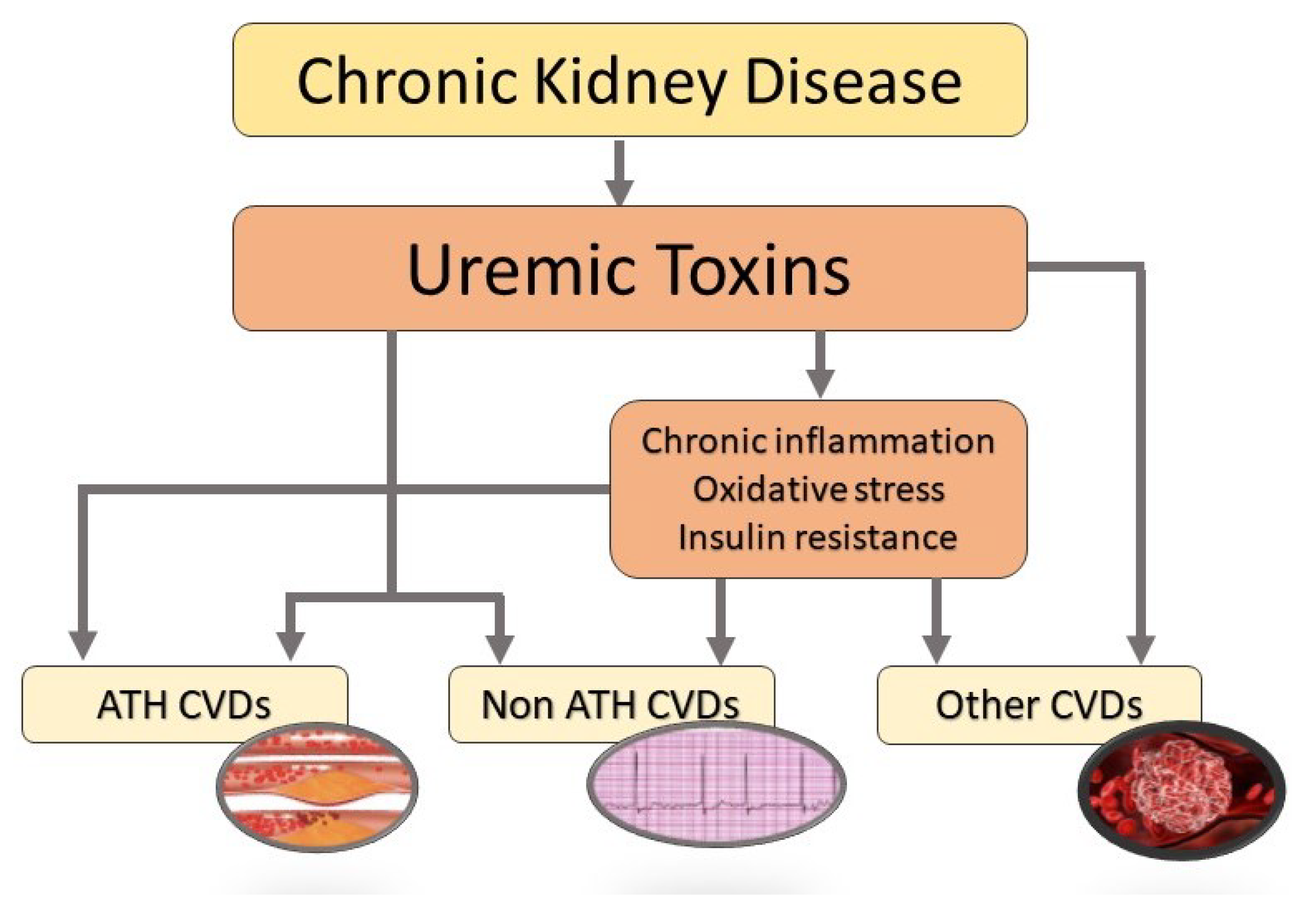
:1. Introduction
2. Cardiovascular Diseases
2.1. Atheromatous Cardiovascular Diseases
2.2. Non-Atheromatous Cardiovascular Diseases
2.3. Other than Atheromatous and Non Atheromatous Cardiovascular Diseases
3. Uremic Toxins and Risk for Cardiovascular Diseases
3.1. Experimental Data: In Vitro Studies

{kind=link}
| First Author, Year | Models | UT(s) Studied | Main Findings |
|---|---|---|---|
| Arinze [50], 2022 | Primary human dermal | IS | IS, kynurenine, and KA decreased Wnt/-catenin |
| microvascular ECs | Kynurenine | activity, which causes EC dysfunction and impairs | |
| KA | angiogenesis. | ||
| Lano [93], 2020 | HUVECs | IS | IS had a prothrombotic effect by increasing TF expression in ECs and peripheral blood mononuclear cells via AHR activation. |
| He [80], 2019 | HASMCs | IS | IS induced calcification of HASMCs via the NF-B signaling pathway. |
| Chen [81], 2016 | HASMCs | IS | IS decreased Klotho expression, promoting aortic calcification. |
| Tang [90], 2015 | Embryonic rat heart-derived cardiac H9c2 cells | IS | IS has a role in arrhythmogenesis: IS inhibited the inward rectifier potassium ion channels function, resulting in a prolonged QT interval. |
| Chitalia [94], 2013 | HVSMCs | IS | IS increased TF expression and decreased TF ubiquitination, leading to a thrombogenic milieu. |
| Liu [92], 2012 | Neonatal cardiac myocytes and fibroblasts from Sprague–Dawley rats | IS | IS was taken up by cardiomyocytes through OAT-1 and -3, leading to activation of the NF-B and MAPK pathways that are involved in cardiac hypertrophy and fibrosis. |
| Lekawanvijit [91], 2010 | Isolated NCMs, NCFs and THP-1 | IS | IS has a role in harmful cardiac remodeling: it has pro-fibrotic, pro-hypertrophic, and pro-inflammatory effects via the activation of MAPK and NF-B pathways. |
| Tumur [62], 2010 and Ito [63], 2010 | HUVECs | IS | IS increased the expression of the adhesion molecules ICAM-1, VCAM-1, MCP-1, and e-selectin, all of which are involved in the pathophysiology of atherosclerosis. |
| Muteliefu [51], 2009 | HASMCs | IS | IS induced ROS generation and the expression of Nox4, Cbfa1, ALP, and osteopontin in VSMCs. |
| Yamamoto [64], 2006 | VSMCs were isolated from the aortas of male Sprague–Dawley rats | IS | IS caused VSMC proliferation via activation of the p42/44 MAPK pathway, a mechanism involved in the progression of atherosclerotic lesions. |
| Dou [52], 2015 | Cultured human endothelial cells | IAA | IAA activated the inflammatory AHR/p38MAPK/NF-B pathway and increased the production of endothelial ROS. |
| Gao [96], 2015 | RBC from peripheral vein | IAA | IS and IAA caused RBC damage, which is involved |
| blood of eight healthy volunteers | IS | in thrombus formation. | |
| Gondouin [95], 2013 | HUVECs | IAA | IAA increased TF expression resulting in a prothrombotic effect. |
| Gross [65], 2015 | HUVECs and HVSMCs | PCS | PCS directly stimulated the Rho-associated protein kinase, which is involved in vascular dysfunction and vascular remodeling. |
| Watanabe [53], 2015 | HUVECs | PCS | PCS enhanced ROS production and NADPH oxidase expression. |
| Meijers [66], 2009 | HUVECs | PCS | PCS induced shedding of endothelial microparticles, causing endothelial dysfunction. |
| Schepers [58], 2007 | Blood from healthy donors incubated in the presence of PCS | PCS | The presence of PCS activated pro-inflammatory leukocyte free radical production. |
| Dou [67], 2004 | HUVECs | PCS | Both PCS and IS inhibited endothelial proliferation |
| IS | and wound repair. | ||
| Huang [60], 2018 | Human aortic endothelial cells | HA | HA contributed to mitochondrial fission by activating mitochondrial ROS production and Drp1 protein expression. |
| Shang [61], 2017 | HUVECs | HA | HA, IS, and IAA increased miR-92a levels, which im- |
| IS | pairs EC function. | ||
| IAA | |||
| Nagy [97], 2017 | Human islets of Langerhans from healthy donors | CMPF | CMPF inhibited insulin secretion. |
| Itoh [59], 2012 | HUVECs | CMPF | IS induced ROS production more intensely than |
| IS | CMPF did. | ||
| Bouabdallah [82], 2019 | HUVECs and HASMCs | Phosphate | Phosphate and IS induced the secretion of interleuk- |
| IS | in-8 from ECs, which is involved in VSMC calcification. | ||
| Jover [83], 2018 | VSMCs | Phosphate | High phosphate promoted extracellular matrix calcification and upregulated osteoblast markers. |
| Zhang [84], 2017 | HASMCs | Phosphate | High phosphate induced vascular calcification via the activation of TLR4/NF-B signaling. |
| Alesutan [85], 2017 | HASMCs | Phosphate | Hyperphosphatemia upregulated aldosterone synthase expression, inducing VSMCs osteogenic transdifferentiation and calcification. |
| Rahabi-Layachi [68], 2015 | HASMCs | Phosphate | Phosphate induced apoptosis and cell cycle arrest by blocking G1/S progression, thus reducing HASMCs proliferation. |
| M’Baya-Moutoula [86], 2015 | Peripheral blood mononuclear cells | Phosphate | Phosphate caused vascular calcification by modulating miR-223 and decreasing osteoclastogenesis. |
| Ciceri [87], 2015 | VSMCs | Phosphate | Phosphate caused VSMC osteoblastic differentiation and led to cell calcification. |
| Di Marco [69], 2013 | Human coronary artery ECs | Phosphate | Hyperphosphatemia decreased annexin II expression and stiffened ECs. |
| Six [70], 2012 | HUVECs | Phosphate | Phosphate exhibited a direct vasoconstrictor effect on aortic rings, increased phenylephrine-induced contraction, and lowered acetylcholine-induced relaxation—leading to endothelial dysfunction. |
| Guerrero [88], 2012 | Rat aortic rings and HVSMCs | Phosphate | Phosphate reduced expression of perlecan and induced BMP-2, which is involved in the osteogenic transdifferentiation pathways and would promote cells calcification. |
| Shroff [89], 2010 | VSMCs | Phosphate | Phosphate increased alkaline phosphatase activity and mediated calcification. |
| Di Marco [54], 2008 | HUVECs | Phosphate | Hyperphosphatemia caused EC apoptosis by increasing ROS generation and disrupting the mitochondrial membrane potential. |
| Shigematsu [71], 2003 | HVSMCs | Phosphate | Phosphate overload accelerated calcium deposition on arteriole walls. Moreover, phosphate led to vasoconstriction, decreased vasorelaxation, decreased NO production, stimulated ROS production, and induced ECs apoptosis. |
| Lee [72], 2021 | HUVECs | Urea | Urea led to excessive neutrophil extracellular trap formation and thus EC dysfunction. |
| Maciel [73], 2018 | An immortalized human EC line | Urea | Urea altered cell-to-cell junctions, leading to greater endothelial damage. |
| D’Apolito [55], 2018 | Human arterial ECs | Urea | Abnormal high urea levels had long-lasting effects on arterial cells: urea increased mitochondrial ROS production in arterial ECs even after dialysis, which typically promotes endothelial dysfunction. |
| D’Apolito [56], 2017 | Human endothelial progenitor cell | Urea | Urea caused ROS production and accelerated endothelial progenitor cell senescence. |
| Sun [75], 2016 | Human arterial EC | Urea | Urea levels were positively correlated with HDL carbamylation, which inhibited endothelial repair functions. |
| D’Apolito [57], 2015 | Human aortic ECs | Urea | Urea increased mitochondrial ROS production and inhibited GAPDH, which leads to the activation of the endothelial pro-inflammatory pathway. Furthermore, urea inactivated the anti-atherosclerosis enzyme PGI2 synthase. |
| Trécherel [74], 2012 | HASMCs | Urea | Urea induced BAD protein expression, sensitizing the HASMCs to apoptosis. |
| D’Apolito [98], 2010 | 3T3-L1 adipocytes treated with urea | Urea | Urea increased ROS levels and expression of the adipokines retinol binding protein 4 and resistin. |
| Zhang [76], 2020 | Aortic VSMCs from male “Sprague Dawley” rats and human VSMCs | TMAO | TMAO promoted vascular calcification through activation of the NLRP3 inflammasome and NF-B signals. |
| Ma [77], 2017 | HUVECs | TMAO | HUVECs showed impairment in cellular proliferation, and TMAO induced NF-B signaling pathway, increasing vascular inflammatory signals and EC dysfunction. |
| Boini [78], 2017 | Mouse carotid artery ECs | TMAO | TMAO activated NLRP3 inflammasomes, causing endothelial dysfunction. |
| Sun [79], 2016 | HUVECs | TMAO | TMAO activated NLRP3 inflammasomes, causing endothelial dysfunction. |
3.2. Experimental Data: Animal Studies
3.2.1. Atheromatous Cardiovascular Diseases
3.2.2. Non-Atheromatous Cardiovascular Diseases
3.2.3. Other than Atheromatous and Non-Atheromatous Cardiovascular Diseases
| First Author, Year | Models | UT(s) Studied | Main Findings |
|---|---|---|---|
| Atheromatous CVDs | |||
| Arinze [50], 2022 | Adenine-induced | IS | IS, kynurenine, and KA suppressed Wnt/- |
| CKD mice and IS so- | Kynurenine | catenin signaling through increased AHR activity, | |
| lute-specific C57BL/6 | KA | leading to impaired angiogenesis and hindlimb | |
| mice | ischemia. | ||
| Hung [101], 2016 | Mice with subtotal nephrectomy | IS | IS decreased endothelial progenitor cells mobilization and impaired neovascularization, leading to PAD. |
| Han [103], 2016 | 5/6 nephrectomized ApoE –/– mice | PCS | PCS promoted the formation of atherosclerotic lesions, induced plaque instability and the migration and proliferation of VSMCs, and disturbed the balance between matrix metalloproteinases and tissue inhibitor of metalloproteinases within the plaques. |
| Huang [60], 2018 | 5/6 nephrectomized rat model | HA | HA caused pro-atherogenic effects by contributing to endothelial dysfunction via greater oxidative stress and impaired endothelium-dependent vasodilation. |
| Shang [61], 2017 | Male Wistar rats | HA | HA induced miR-92a, which is involved in angiogenic and atherosclerotic processes. |
| Massy [107], 2005 | ApoE −/− mice with partial kidney ablation | Urea | Urea contributed to arterial calcification and aggravated atherosclerosis. |
| Matsumoto [102], 2020 | Superior mesenteric arteries and femoral arteries of rat | TMAO | TMAO impaired endothelium-derived hyperpolarizing factor-type relaxation, which led to PAD. |
| Geng [104], 2018 | Apoe −/− mice fed a high-fat diet with or without TMAO | TMAO | TMAO enhanced the expression of CD36/MAPK/JNK pathway, promoting foam cells formation and, ultimately, atherosclerosis. |
| Seldin [105], 2016 | Female low-density lipoprotein receptor knockout mice injected with vehicle or TMAO | TMAO | TMAO induced vascular inflammation by activating MAPK and NF-B signaling, thus enhancing atherosclerosis. |
| Koeth [106], 2013 | Mice supplemented with dietary TMAO, carnitine, or choline | TMAO | TMAO accelerated atherosclerosis and was linked to major cardiac events. |
| Non-atheromatous CVDs | |||
| Kuo [108], 2020 | Nephrectomized male C57BL/6 mice | IS | IS promoted calcification in the aorta and peripheral arteries, with low NO production and high eNOS phosphorylation. |
| Opdebeeck [109], 2019 | 42 male Wistar rats ex- | IS | Both IS and PCS directly promoted severe calcifica- |
| posed to adenine sulfate for 10 days and then fed a phosphate-enriched diet | PCS | tion in the aorta and peripheral vessels via activation of inflammation and coagulation pathways. These changes were strongly associated with impaired glucose homeostasis. | |
| Chen [81], 2016 | 5/6 nephrectomized Sprague Dawley rats treated with IS | IS | IS decreased Klotho expression and promoted aortic calcification. |
| Chen [121], 2015 | Isolated rabbit left atrium, right atrium, pulmonary vein, and sinoatrial nodes before and after treatment with IS | IS | IS may contribute to atrial fibrillation: It increased pulmonary vein and atrial arrhythmogenesis through oxidative stress, inflammation, and fibrosis. |
| Yisireyili [119], 2013 and Lekawanvijit [120], 2012 | Dahl salt-sensitive hypertensive rats | IS | IS aggravated cardiac fibrosis and cardiomyocyte hypertrophy, with greater levels of oxidative stress and lower anti-oxidative defenses. |
| Muteliefu [110], 2012 | Aorta of subtotally nephrectomized Dahl salt-sensitive hypertensive rats | IS | IS accelerated VSMC senescence and vascular calcification, with upregulation of p21, p53, and prelamin A through oxidative stress. |
| Adijiang [111], 2010 | Dahl salt-sensitive hypertensive rats | IS | IS increased aortic calcification and wall thickness; induced expression of p16, p21, p53 and Rb in the calcification area; and thus promoted cell senescence. |
| Adijiang [112], 2008 | Dahl salt-sensitive hypertensive rats | IS | IS induced aortic calcification (with expression of osteoblast-specific proteins) and aortic wall thickening. |
| Han [126], 2015 | 5/6 nephrectomized mice | PCS | PCS promoted cardiac apoptosis and diastolic dysfunction by upregulating the expression of NADPH oxidase and the production of ROS. |
| Hu [123], 2015 | Two CKD rodent models: UNX-IRI26 and 5/6 nephrectomized | Phosphate | High phosphate was associated with lower Klotho levels, leading to cardiac hypertrophy and fibrosis. |
| Yamada [116], 2014 | Adenine-induced CKD male Sprague–Dawley rats | Phosphate | High phosphate directly increased the expression of TNF- and osteochondrogenic markers, inducing systemic inflammation and vascular calcification. |
| Lau [114], 2013 | DBA/2 mice with partial renal ablation | Phosphate | High phosphate was associated with arterial medial calcification. |
| Crouthamel [113], 2013 | Mice with targeted deletion of PiT-1 in VSMCs | Phosphate | High phosphate induced calcification of VSMCs. |
| El-Abbadi [115], 2009 | Female DBA/2 mice induced uremia with left total nephrectomy | Phosphate | High phosphate was associated with extensive arterial medial calcification. |
| Graciolli [117], 2009 | 5/6 nephrectomized Wistar rats with parathyroidectomy | Phosphate | Phosphate upregulated aortic expression of Runx2 and led to calcified VSMC. |
| Hosaka [118], 2009 | 5/6 nephrectomized male Sprague-Dawley rats | Phosphate | High phosphate induced elastin degradation via the upregulation of tissue-nonspecific alkaline phosphatase, accelerating the transformation of VSMCs into osteoblast-like cells and leading to medial layer calcification. |
| Zhu [122], 2021 | 25 nephrectomized SPF-grade male Sprague–Dawley rats | Urea | Urea caused myocardial hypertrophy. |
| Prommer [124], 2018 | 11 uremic mice and 8 controls | Urea | Urea led to systemic microvascular disease, with microvascular rarefaction, tissue hypoxia, and dysfunctional angiogenesis. |
| Carmona [125], 2011 | 2 groups of 30 Wistar male rats: 1 with renal ablation and the other with kidney manipulation only | Urea | Urea induced systemic inflammation and led to the thickening of subepicardiac arteries. |
| Other than ATH and non ATH CVDs | |||
| Yang [127], 2017 | C57BL/6J mice with left total nephrectomy | IS | IS activated ROS/p38 MAPK signaling and reduced Klotho expression, which induced platelet aggregation and thrombus formation. |
| Kolachalama [128], 2018 | A group of C57BL/6 mice administered Kyn, the excretion of which was inhibited by probenecid | Kynurenine | High kynurenine levels promoted clotting in response to vascular injury. |
| Koppe [133], 2017 | 5/6 nephrectomized | PCS | PCS (but not PCG) promoted insulin resistance. |
| mice | PCG | ||
| Koppe [134], 2013 | CD1 Swiss and C57BL/6J mice with 5/6 nephrectomy | PCS | PCS contributed to insulin resistance: It altered insulin signaling in skeletal muscle through the activation of extracellular signal-regulated kinases. |
| Nagy [97], 2017 | Male CD1 mice injected with CMPF | CMPF | CMPF inhibited insulin secretion. |
| Koppe [130], 2016 | C57BL/6N male mice with 5/6 nephrectomy | Urea | Urea increased oxidative stress and protein O-GlcNAcylation, impairing insulin secretion and glycolysis. |
| Carracedo [131], 2013 | 5/6 nephrectomized 40 male Wistar rats | Urea | Urea induced oxidative stress, leading to EC damage. |
| D’Apolito [98], 2010 | 5/6 nephrectomized C57BL/6J wild-type mice | Urea | Urea increased ROS production and induced insulin resistance and glucose intolerance. |
| Li [132], 2018 | 5/6 nephrectomized rats | TMAO | High TMAO levels decreased NO production, contributing to endothelial dysfunction. |
| Zhu [129], 2016 | Carotid artery thrombosis models of germ-free C57BL/6J female mice | TMAO | TMAO enhanced submaximal stimulus-dependent platelet activation, increasing the thrombosis risk. |
3.3. Observational Studies
3.3.1. Atheromatous Cardiovascular Diseases
3.3.2. Non-Atheromatous Cardiovascular Diseases
3.3.3. Other than Atheromatous and Non-Atheromatous Cardiovascular Diseases
| First Author, Year | Models | UT(s) Studied | Main Findings |
|---|---|---|---|
| Atheromatous CVDs | |||
| Arinze [50], 2022 | 20 HD patients and 15 | IS | Elevated plasma levels of IS, kynurenine, and KA in |
| controls | Kynurenine | HD patients showed a significant decrease in ECs | |
| KA | proliferation and migration, compared with the control group. | ||
| Arinze [50], 2022 | PAD patients: 35 without | IS | Elevated plasma levels of IS, kynurenine, KA, with |
| adverse limb event and | Kynurenine | suppressed Wnt activity in ECs were associated with | |
| 28 with | KA | an increased risk of future adverse limb events. | |
| Shafi et al. [153], 2015 | 394 incident HD patients | IS | Elevated serum levels of IS, PCS, PAG and HA were |
| PCS | associated with greater risk of fatal or nonfatal | ||
| PAG | atherosclerotic cardiovascular events in incident HD | ||
| HA | patients. | ||
| Hsu [154], 2013 | 191 mild-to-moderate CKD patients | IS | Elevated serum IS levels were associated with coronary atherosclerosis and correlated with the severity of the disease. |
| Melamed [167], 2013 | 521 incident HD patients | IS | IS and PCS were not associated with atherosclerotic |
| PCS | cardiovascular death. | ||
| Lin [155], 2012 | 70 pre-dialysis patients (CKD stage 3 to 5) | IS | Serum IS levels were positively correlated with atherosclerotic cardiovascular events. |
| Lin [152], 2012 | 100 stable HD patients | IS | Elevated serum levels of IS and PCS were associated |
| PCS | with PAD and arteriosclerosis markers. | ||
| Lin [157], 2010 | 100 HD patients | IS | Only elevated serum PCS levels were significantly |
| PCS | associated with fatal or nonfatal atherosclerotic cardiovascular events. | ||
| Taki [156], 2007 | 224 HD patients | IS | Plasma IS levels were significantly and negatively correlated with HDL cholesterol and were positively associated with atherosclerotic lesions. |
| Poesen [158], 2016 | 488 patients (all CKD | PCS | A lower serum PCS:PCG ratio and a higher |
| stages) | PCG | total PCS + PCG level were associated with fatal or nonfatal atherosclerotic CVDs. | |
| Wang [160], 2010 | 202 patients with stable angina and early-stage kidney failure | PCS | Elevated plasma PCS levels were associated with coronary artery disease and correlated with the severity of the disease. |
| Poesen [159], 2016 | 488 patients with CKD stages 1–5 | PAG | An elevated serum PAG level was a powerful, independent risk factor for major CVD (such as MI and stroke). |
| Merhi [162], 2017 | 3138 CKD patients | Phosphate | Hyperphosphatemia was associated with atherosclerotic CVD. |
| Eddington [163], 2010 | 1203 nondialyzed CKD patients | Phosphate | Hyperphosphatemia increased the risk of cardiovascular death from atheromatous CVD. |
| Kestenbaum [164], 2005 | 3490 CKD patients | Phosphate | Hyperphosphatemia was associated with MI. |
| Nakamura [161], 2002 | 525 HD patients | Phosphate | Hyperphosphatemia was associated with atherosclerotic diseases. |
| Stubbs [165], 2016 | 104 CKD patients | TMAO | Elevated TMAO concentrations were correlated with coronary atherosclerosis. |
| Kim [166], 2016 | 2529 patients (stages 3b and 4 CKD) | TMAO | Elevated serum TMAO levels were associated with ischemic cardiovascular events. |
| Non-atheromatous CVDs | |||
| Chinnappa [168], 2018 | 56 male patients with | IS | These serum UT levels showed significant negative |
| stage 2–5 CKD, nondia- | IAA | correlation with peak cardiac power and subclinical | |
| lyzed and free of heart | PCS | cardiac dysfunction, but no correlation with left ven- | |
| disease | PCG | tricular mass index was found. | |
| HA | |||
| Cao [169], 2015 | 258 HD patients | IS | Elevated plasma IS was associated with heart failure. |
| Sato [171], 2013 | 204 CKD patients with preserved left ventricular function | IS | Elevated plasma IS levels were associated with an increased risk of left ventricular diastolic dysfunction. |
| Shimazu [170], 2013 | 76 patients with mild-to-moderate CKD and dilated cardiomyopathy | IS | Elevated serum IS levels were associated with hospitalization for heart failure and cardiac death. |
| Barreto [172], 2009 | 139 patients with CKD from stage 2 to dialysis | IS | Being in the highest serum IS tertile was directly associated with pulse wave velocity, aortic calcification, and higher cardiovascular mortality. |
| Zapolski [173], 2020 | 100 CKD patients with persistent atrial fibrillation | KA | Serum KA levels were positively correlated with aortic stiffness and indices of diastolic dysfunction of left atrium and left ventricle. |
| Pawlak [174], 2010 | 106 CKD patients | KA | Elevated plasma kynurenine and KA levels were as- |
| Kynurenine | sociated with intima-media thickness. | ||
| Liabeuf [175], 2010 | 139 CKD patients | PCS | Elevated total and free serum PCS levels were significantly associated with vascular calcification, and free PCS was shown to be a predictor of cardiovascular death. |
| Yu [176], 2018 | 80 HD patients | HA | Elevated HA levels were significantly associated with left ventricular hypertrophy. |
| Petchey [177], 2012 | 120 CKD pre-dialysis patients | Phosphate | Serum phosphate was positively correlated with aortic pulse wave velocity, arterial stiffness, and the presence of vascular calcification. |
| Adeney [178], 2009 | 6814 patients with CKD aged 45–84 | Phosphate | Hyperphosphatemia was associated with vascular and valvular calcification. |
| Ix [180], 2009 | 440 patients with moderate CKD | Phosphate | Hyperphosphatemia was strongly associated with peripheral arterial stiffness. |
| Ketteler [179], 2003 | 312 HD patients | Phosphate | Hyperphosphatemia was associated with vascular calcification and cardiovascular mortality. |
| Drechsler [181], 2015 | 1255 HD patients | Urea | Higher blood urea levels were associated with higher tertile serum carbamylated albumin levels, which in turn were positively correlated with heart failure and arrhythmia. |
| Atheromatous and non-atheromatous CVDs | |||
| Chen [147], 2021 | 3407 participants with | IS | Lower 24-h kidney clearance of IS, KA, and PCS |
| CKD, excluding those | KA | was not found to be associated with heart failure and | |
| with a GFR < 20 mL/min/1.73 m2 | PCS | MI after adjustment for GFR. | |
| Fan [135], 2019 | 147 patients with CKD stage 1–5 | IS | Elevated plasma IS levels were associated with major adverse cardiovascular events, independently of GFR and nutritional status. |
| Shafi [148], 2017 | 1273 HD patients | IS | Overall, elevated serum IS, PCS, PAG and HA levels |
| PCS | were not associated with any cardiovascular event. | ||
| PAG | However, high IS levels were predictive of cardiac | ||
| HA | and sudden cardiac death in patients with low albumin levels. | ||
| Konje [136], 2021 | 92 CKD patients with a history of CVD, 46 with no history of CVD, and 46 with incident CVD | Kynurenine | Elevated serum kynurenine levels were associated with incident atheromatous and non-atheromatous CVDs. |
| Wu [137], 2012 | 112 HD patients aged from 65 to 90 | PCS | Elevated free PCS serum levels were associated with cardiovascular mortality. |
| Liabeuf [138], 2013 | 139 CKD patients | PCG | Elevated free and total serum PCG levels were correlated with cardiovascular mortality independently of survival predictors. |
| Luce [149], 2018 | 270 HD patients | CMPF | Elevated serum CMPF was not associated with any CVD. |
| McGovern [139], 2013 | 13,292 CKD patients at stages 3–5 | Phosphate | Hyperphosphatemia was correlated with increased CVDs. |
| Kimata [140], 2007 | 3973 HD patients | Phosphate | Hyperphosphatemia was significantly associated with cardiovascular mortality. |
| Menon [150], 2005 | 840 CKD patients | Phosphate | Hyperphosphatemia was significantly associated with increased cardiovascular mortality but only before adjustment for GFR. |
| Slinin [141], 2005 | 14829 HD patients | Phosphate | Hyperphosphatemia was associated with CVDs and mortality. |
| Young [142], 2005 | 17236 dialysis patients | Phosphate | Hyperphosphatemia was significantly associated with cardiovascular mortality. |
| Block [143], 2004 | 40538 HD patients | Phosphate | Hyperphosphatemia was significantly associated with cardiovascular hospitalization and mortality. |
| Laville [27], 2022 | 2507 CKD patients before RRT | Urea | Higher serum urea levels were associated with a greater risk of CVD. |
| Berg [146], 2013 | 187 HD patients | Urea | Urea was positively correlated with carbamylation of serum albumin, which is associated with CVDs and mortality. |
| Shafi [144], 2017 | 1846 prevalent HD patients | TMAO | An elevated serum TMAO concentration was associated with cardiovascular events and death. |
| Kaysen [151], 2015 | 235 HD patients | TMAO | There was no significant association between TMAO and cardiovascular hospitalizations or death. |
| Other than ATH and non ATH CVDs | |||
| Glorieux [182], 2021 | 523 nondialyzed patients | IS | Elevated serum levels of these UTs were correlated |
| (all stages of CKD) | IAA | with markers of endothelial damage (mainly angio- | |
| PCS | poietin-2). Elevated levels of free PCS and free PCG | ||
| PCG | had the strongest association with CVD, indepen- | ||
| HA | dently of the GFR. | ||
| Wang [184], 2019 | 110 patients with stage 3–5 CKD | IS | Elevated levels of serum IS were negatively correlated with vascular reactivity index values, leading to endothelial dysfunction. |
| Kolachalama [128], 2018 | 473 participants under- | IS | Elevated serum levels of IS and kynurenine were |
| going angioplasty for dialysis access dysfunction | Kynurenine | associated with postangioplasty thrombosis of dialysis grafts. | |
| Wu [190], 2016 | 306 patients undergoing angioplasty for dialysis access dysfunction | IS | Elevated serum levels of IS were associated with postangioplasty thrombosis of dialysis grafts. |
| Jourde-Chiche [185], 2009 | 38 HD patients and 21 | IS | Elevated serum levels of IS, IAA, and PCS were asso- |
| healthy controls | IAA | ciated with low numbers of endothelial progenitor | |
| PCS | cells. | ||
| Pawlak [187], 2010 | 64 patients on peritoneal dialysis | KA | Plasma KA levels were positively associated with TF inhibitor and negatively associated with prothrombin fragment 1 + 2 levels. |
| Pawlak [188], 2009 | 48 patients with ESRD | Kynurenine | Plasma kynurenine levels were positively associated with thrombomodulin and von Willebrand factor (markers of endothelial dysfunction). |
| Pawlak [186], 2009 | 146 CKD patients with 91 | Kynurenine | Elevated serum levels of kynurenine and KA were |
| ones on dialysis | KA | associated with increased oxidative stress, inflammation, and endothelial dysfunction. | |
| Pawlak [189], 2009 | 92 patients on dialysis | Kynurenine | Elevated serum levels of kynurenine and KA were |
| KA | independently and significantly associated with hypercoagulability. | ||
| Meijers [66], 2009 | 100 HD patients | PCS | Elevated serum PCS levels were associated with the levels of circulating endothelial microparticles. |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perico, N.; Remuzzi, G. Chronic kidney disease: A research and public health priority. Nephrol. Dial. Transplant. 2012, 27, iii19–iii26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levey, A.S.; Eckardt, K.U.; Dorman, N.M.; Christiansen, S.L.; Hoorn, E.J.; Ingelfinger, J.R.; Inker, L.A.; Levin, A.; Mehrotra, R.; Palevsky, P.M.; et al. Nomenclature for kidney function and disease: Report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int. 2020, 97, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.C.; Zhang, L.X. Prevalence and disease burden of chronic kidney disease. Ren. Fibros. Mech. Ther. 2019, 1165, 3–15. [Google Scholar]
- Nlandu, Y.; Padden, M.; Seidowsky, A.; Hamaz, S.; Vilaine, E.; Cheddani, L.; Essig, M.; Massy, Z.A. Toxines urémiques de moyen poids moléculaire: Un véritable regain d’intérêt. Néphrol. Thér. 2019, 15, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszek, E.; Oldakowska-Jedynak, U.; Kwiatkowska, M.; Glogowski, T.; Malyszko, J. Uremic toxins, oxidative stress, atherosclerosis in chronic kidney disease, and kidney transplantation. Oxidative Med. Cell. Longev. 2021, 2021, 6651367. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.L.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Drüeke, T.B.; Massy, Z.A. Atherosclerosis in CKD: Differences from the general population. Nat. Rev. Nephrol. 2010, 6, 723–735. [Google Scholar] [CrossRef]
- Villain, C.; Metzger, M.; Combe, C.; Fouque, D.; Frimat, L.; Jacquelinet, C.; Laville, M.; Briançon, S.; Klein, J.; Schanstra, J.P.; et al. Prevalence of atheromatous and non-atheromatous cardiovascular disease by age in chronic kidney disease. Nephrol. Dial. Transplant. 2020, 35, 827–836. [Google Scholar] [CrossRef] [Green Version]
- Liabeuf, S.; Drüeke, T.B.; Massy, Z.A. Protein-bound uremic toxins: New insight from clinical studies. Toxins 2011, 3, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; De Smet, R.; Glorieux, G. Review on uremic toxins: Classification, concentration, and interindividual variability (volume 63, pg 1934, 2003). Kidney Int. 2020, 98, 1354. [Google Scholar]
- Rosner, M.H.; Reis, T.; Husain-Syed, F.; Vanholder, R.; Hutchison, C.; Stenvinkel, P.; Blankestijn, P.J.; Cozzolino, M.; Juillard, L.; Kashani, K.; et al. Classification of uremic toxins and their role in kidney failure. Clin. J. Am. Soc. Nephrol. 2021, 16, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graboski, A.L.; Redinbo, M.R. Gut-derived protein-bound uremic toxins. Toxins 2020, 12, 590. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J.; European Uremic Toxin Work Group. A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradi, H.; Sica, D.A.; Kalantar-Zadeh, K. Cardiovascular burden associated with uremic toxins in patients with chronic kidney disease. Am. J. Nephrol. 2013, 38, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Pieniazek, A.; Bernasinska-Slomczewska, J.; Gwozdzinski, L. Uremic Toxins and Their Relation with Oxidative Stress Induced in Patients with CKD. Int. J. Mol. Sci. 2021, 22, 6196. [Google Scholar] [CrossRef]
- Gross, P.; Six, I.; Kamel, S.; Massy, Z.A. Vascular Toxicity of Phosphate in Chronic Kidney Disease–Beyond Vascular Calcification. Circ. J. 2014, 78, 2339–2346. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhao, Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.J.; Sidor, N.A.; Tonial, N.C.; Che, A.; Urquhart, B.L. Uremic Toxins in the Progression of Chronic Kidney Disease and Cardiovascular Disease: Mechanisms and Therapeutic Targets. Toxins 2021, 13, 142. [Google Scholar] [CrossRef]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef]
- Association, A.H.; Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; et al. Heart Disease and Stroke Statistics-2020 Update. 2020. Available online: https://www.ahajournals.org/doi/10.1161/CIR.0000000000000757 (accessed on 27 February 2022).
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular disease in chronic kidney disease: Pathophysiological insights and therapeutic options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- London, G.M.; Marchais, S.J.; Guerin, A.P.; Metivier, F.; Adda, H. Arterial structure and function in end-stage renal disease. Nephrol. Dial. Transplant. 2002, 17, 1713–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, U.; Buzello, M.; Ritz, E.; Stein, G.; Raabe, G.; Wiest, G.; Mall, G.; Amann, K. Morphology of coronary atherosclerotic lesions in patients with end-stage renal failure. Nephrol. Dial. Transplant. 2000, 15, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzog, C.A.; Asinger, R.W.; Berger, A.K.; Charytan, D.M.; Díez, J.; Hart, R.G.; Eckardt, K.U.; Kasiske, B.L.; McCullough, P.A.; Passman, R.S.; et al. Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2011, 80, 572–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, K.A.; Mahaffey, K.W.; Mehran, R.; Nissen, S.E.; Wiviott, S.D.; Dunn, B.; Solomon, S.D.; Marler, J.R.; Teerlink, J.R.; Farb, A.; et al. 2017 cardiovascular and stroke endpoint definitions for clinical trials. Circulation 2018, 137, 961–972. [Google Scholar] [CrossRef]
- Laville, S.M.; Couturier, A.; Lambert, O.; Metzger, M.; Mansencal, N.; Jacquelinet, C.; Laville, M.; Frimat, L.; Fouque, D.; Combe, C.; et al. Urea levels and cardiovascular disease in patients with chronic kidney disease. Nephrol. Dial. Transplant. 2022, gfac045. [Google Scholar] [CrossRef]
- Valdivielso, J.M.; Rodríguez-Puyol, D.; Pascual, J.; Barrios, C.; Bermúdez-López, M.; Sánchez-Niño, M.D.; Pérez-Fernández, M.; Ortiz, A. Atherosclerosis in chronic kidney disease: More, less, or just different? Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1938–1966. [Google Scholar] [CrossRef]
- Masson, P.; Webster, A.C.; Hong, M.; Turner, R.; Lindley, R.I.; Craig, J.C. Chronic kidney disease and the risk of stroke: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2015, 30, 1162–1169. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Mohler, E.R.; Xie, D.; Shlipak, M.; Townsend, R.R.; Appel, L.J.; Ojo, A.; Schreiber, M.; Nessel, L.; Zhang, X.; et al. Traditional and non-traditional risk factors for incident peripheral arterial disease among patients with chronic kidney disease. Nephrol. Dial. Transplant. 2016, 31, 1145–1151. [Google Scholar] [CrossRef]
- Wattanakit, K.; Folsom, A.R.; Selvin, E.; Coresh, J.; Hirsch, A.T.; Weatherley, B.D. Kidney function and risk of peripheral arterial disease: Results from the Atherosclerosis Risk in Communities (ARIC) Study. J. Am. Soc. Nephrol. 2007, 18, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Odutayo, A.; Wong, C.X.; Hsiao, A.J.; Hopewell, S.; Altman, D.G.; Emdin, C.A. Atrial fibrillation and risks of cardiovascular disease, renal disease, and death: Systematic review and meta-analysis. BMJ 2016, 354, i4482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverberg, D.; Wexler, D.; Blum, M.; Schwartz, D.; Iaina, A. The association between congestive heart failure and chronic renal disease. Curr. Opin. Nephrol. Hypertens. 2004, 13, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, P.A.; Loutradis, C.; Karpetas, A.; Tzanis, G.; Piperidou, A.; Koutroumpas, G.; Raptis, V.; Syrgkanis, C.; Liakopoulos, V.; Efstratiadis, G.; et al. Ambulatory pulse wave velocity is a stronger predictor of cardiovascular events and all-cause mortality than office and ambulatory blood pressure in hemodialysis patients. Hypertension 2017, 70, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Briet, M.; Bozec, E.; Laurent, S.; Fassot, C.; London, G.; Jacquot, C.; Froissart, M.; Houillier, P.; Boutouyrie, P. Arterial stiffness and enlargement in mild-to-moderate chronic kidney disease. Kidney Int. 2006, 69, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Pannier, B.; Guérin, A.P.; Marchais, S.J.; Safar, M.E.; London, G.M. Stiffness of capacitive and conduit arteries: Prognostic significance for end-stage renal disease patients. Hypertension 2005, 45, 592–596. [Google Scholar] [CrossRef] [Green Version]
- London, G.M. Arterial stiffness in chronic kidney disease and end-stage renal disease. Blood Purif. 2018, 45, 154–158. [Google Scholar] [CrossRef]
- Lee, S.J.; Lee, I.K.; Jeon, J.H. Vascular calcification—new insights into its mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef] [Green Version]
- Vahed, S.Z.; Mostafavi, S.; Khatibi, S.M.H.; Shoja, M.M.; Ardalan, M. Vascular calcification: An important understanding in nephrology. Vasc. Health Risk Manag. 2020, 16, 167. [Google Scholar] [CrossRef]
- Ozcan, C. Conduction Intervals and Atrial Fibrillation in Chronic Kidney Disease. Am. J. Nephrol. 2021, 52, 354–355. [Google Scholar] [CrossRef]
- Kaya, B.; Paydas, S.; Aikimbaev, K.; Altun, E.; Balal, M.; Deniz, A.; Kaypakli, O.; Demirtas, M. Prevalence of cardiac arrhythmia and risk factors in chronic kidney disease patients. Saudi J. Kidney Dis. Transplant. 2018, 29, 567. [Google Scholar] [CrossRef]
- Bonato, F.O.B.; Canziani, M.E.F. Ventricular arrhythmia in chronic kidney disease patients. Braz. J. Nephrol. 2017, 39, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and oxidative stress in chronic kidney disease—potential therapeutic role of minerals, vitamins and plant-derived metabolites. Int. J. Mol. Sci. 2020, 21, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gremmel, T.; Müller, M.; Steiner, S.; Seidinger, D.; Koppensteiner, R.; Kopp, C.W.; Panzer, S. Chronic kidney disease is associated with increased platelet activation and poor response to antiplatelet therapy. Nephrol. Dial. Transplant. 2013, 28, 2116–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, N.; Wan, F.; Kothari, M.; Adelodun, A.; Ware, J.; Sarode, R.; Hedayati, S.S. Association of platelet function with depression and its treatment with sertraline in patients with chronic kidney disease: Analysis of a randomized trial. BMC Nephrol. 2019, 20, 395. [Google Scholar] [CrossRef] [PubMed]
- Ocak, G.; Verduijn, M.; Vossen, C.Y.; Lijfering, W.M.; Dekker, F.W.; Rosendaal, F.R.; Gansevoort, R.T.; Mahmoodi, B.K. Chronic kidney disease stages 1–3 increase the risk of venous thrombosis. J. Thromb. Haemost. 2010, 8, 2428–2435. [Google Scholar] [CrossRef] [PubMed]
- Folsom, A.R.; Lutsey, P.L.; Astor, B.C.; Wattanakit, K.; Heckbert, S.R.; Cushman, M.; Atherosclerosis Risk in Communities Study. Chronic kidney disease and venous thromboembolism: A prospective study. Nephrol. Dial. Transplant. 2010, 25, 3296–3301. [Google Scholar] [CrossRef]
- Kimura, T.; Morimoto, T.; Kozuma, K.; Honda, Y.; Kume, T.; Aizawa, T.; Mitsudo, K.; Miyazaki, S.; Yamaguchi, T.; Hiyoshi, E.; et al. Comparisons of baseline demographics, clinical presentation, and long-term outcome among patients with early, late, and very late stent thrombosis of sirolimus-eluting stents: Observations from the Registry of Stent Thrombosis for Review and Reevaluation (RESTART). Circulation 2010, 122, 52–61. [Google Scholar]
- Ting, H.H.; Tahirkheli, N.K.; Berger, P.B.; McCarthy, J.T.; Timimi, F.K.; Mathew, V.; Rihal, C.S.; Hasdai, D.; Holmes, D.R., Jr. Evaluation of long-term survival after successful percutaneous coronary intervention among patients with chronic renal failure. Am. J. Cardiol. 2001, 87, 630–633. [Google Scholar] [CrossRef]
- Arinze, N.V.; Yin, W.; Lotfollahzadeh, S.; Napoleon, M.A.; Richards, S.; Walker, J.A.; Belghasem, M.; Ravid, J.D.; Kamel, M.H.; Whelan, S.A.; et al. Tryptophan metabolites suppress Wnt pathway and promote adverse limb events in CKD patients. J. Clin. Investig. 2022, 132, e142260. [Google Scholar] [CrossRef]
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Enoki, Y.; Ishima, Y.; Kadowaki, D.; Kotani, S.; Nakajima, M.; Tanaka, M.; Matsushita, K.; Mori, Y.; et al. p-Cresyl sulfate, a uremic toxin, causes vascular endothelial and smooth muscle cell damages by inducing oxidative stress. Pharmacol. Res. Perspect. 2015, 3, e00092. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Hausberg, M.; Hillebrand, U.; Rustemeyer, P.; Wittkowski, W.; Lang, D.; Pavenstadt, H. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am. J. Physiol. Ren. Physiol. 2008, 294, F1381–F1387. [Google Scholar] [CrossRef] [PubMed]
- D’Apolito, M.; Colia, A.L.; Manca, E.; Pettoello-Mantovani, M.; Sacco, M.; Maffione, A.B.; Brownlee, M.; Giardino, I. Urea memory: Transient cell exposure to urea causes persistent mitochondrial ros production and endothelial dysfunction. Toxins 2018, 10, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Apolito, M.; Colia, A.L.; Lasalvia, M.; Capozzi, V.; Falcone, M.P.; Pettoello-Mantovani, M.; Brownlee, M.; Maffione, A.B.; Giardino, I. Urea-induced ROS accelerate senescence in endothelial progenitor cells. Atherosclerosis 2017, 263, 127–136. [Google Scholar] [CrossRef]
- D’Apolito, M.; Du, X.; Pisanelli, D.; Pettoello-Mantovani, M.; Campanozzi, A.; Giacco, F.; Maffione, A.B.; Colia, A.L.; Brownlee, M.; Giardino, I. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis 2015, 239, 393–400. [Google Scholar] [CrossRef] [Green Version]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main in vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef]
- Itoh, Y.; Ezawa, A.; Kikuchi, K.; Tsuruta, Y.; Niwa, T. Protein-bound uremic toxins in hemodialysis patients measured by liquid chromatography/tandem mass spectrometry and their effects on endothelial ROS production. Anal. Bioanal. Chem. 2012, 403, 1841–1850. [Google Scholar] [CrossRef]
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox Biol. 2018, 16, 303–313. [Google Scholar] [CrossRef]
- Shang, F.; Wang, S.C.; Hsu, C.Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.C.; Wang, Y.T.; Wu, G.; Chien, S.; et al. MicroRNA-92a mediates endothelial dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261. [Google Scholar] [CrossRef] [Green Version]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kB activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, P.; Massy, Z.A.; Henaut, L.; Boudot, C.; Cagnard, J.; March, C.; Kamel, S.; Drueke, T.B.; Six, I. Para-cresyl sulfate acutely impairs vascular reactivity and induces vascular remodeling. J. Cell. Physiol. 2015, 230, 2927–2935. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Rahabi-Layachi, H.; Ourouda, R.; Boullier, A.; Massy, Z.A.; Amant, C. Distinct effects of inorganic phosphate on cell cycle and apoptosis in human vascular smooth muscle cells. J. Cell. Physiol. 2015, 230, 347–355. [Google Scholar] [CrossRef]
- Di Marco, G.S.; König, M.; Stock, C.; Wiesinger, A.; Hillebrand, U.; Reiermann, S.; Reuter, S.; Amler, S.; Köhler, G.; Buck, F.; et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2013, 83, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Maziere, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, T.; Kono, T.; Satoh, K.; Yokoyama, K.; Yoshida, T.; Hosoya, T.; Shirai, K. Phosphate overload accelerates vascular calcium deposition in end-stage renal disease patients. Nephrol. Dial. Transplant. 2003, 18, iii86–iii89. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Nizet, V.; An, J.N.; Lee, H.S.; Song, Y.R.; Kim, S.G.; Kim, J.K. Uremic serum damages endothelium by provoking excessive neutrophil extracellular trap formation. Sci. Rep. 2021, 11, 593. [Google Scholar]
- Maciel, R.A.; Cunha, R.S.; Busato, V.; Franco, C.R.; Gregório, P.C.; Dolenga, C.J.; Nakao, L.S.; Massy, Z.A.; Boullier, A.; Pecoits-Filho, R.; et al. Uremia impacts VE-cadherin and ZO-1 expression in human endothelial cell-to-cell junctions. Toxins 2018, 10, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trécherel, E.; Godin, C.; Louandre, C.; Benchitrit, J.; Poirot, S.; Mazière, J.C.; Massy, Z.A.; Galmiche, A. Upregulation of BAD, a pro-apoptotic protein of the BCL2 family, in vascular smooth muscle cells exposed to uremic conditions. Biochem. Biophys. Res. Commun. 2012, 417, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.T.; Yang, K.; Lu, L.; Zhu, Z.B.; Zhu, J.Z.; Ni, J.W.; Han, H.; Chen, N.; Zhang, R.Y. Increased carbamylation level of HDL in end-stage renal disease: Carbamylated-HDL attenuated endothelial cell function. Am. J.-Physiol. Ren. Physiol. 2016, 310, F511–F517. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C.; et al. Trimethylamine-N-Oxide promotes vascular calcification through activation of NLRP3 (nucleotide-binding domain, Leucine-Rich-Containing family, pyrin Domain-Containing-3) inflammasome and NF-κB (nuclear factor κB) signals. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 751–765. [Google Scholar] [CrossRef]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 2017, 37. Available online: https://pdfs.semanticscholar.org/8ce6/2000e849c3249c7791eb0006a8cadc71d7dd.pdf (accessed on 18 March 2022). [CrossRef] [Green Version]
- Boini, K.M.; Hussain, T.; Li, P.L.; Koka, S.S. Trimethylamine-N-oxide instigates NLRP3 inflammasome activation and endothelial dysfunction. Cell. Physiol. Biochem. 2017, 44, 152–162. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef]
- He, X.; Jiang, H.; Gao, F.; Liang, S.; Wei, M.; Chen, L. Indoxyl sulfate-induced calcification of vascular smooth muscle cells via the PI3K/Akt/NF-κB signaling pathway. Microsc. Res. Tech. 2019, 82, 2000–2006. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Zhang, H.; Liu, T.; Zhang, H.; Teng, J.; Ji, J.; Ding, X. Indoxyl sulfate enhance the hypermethylation of klotho and promote the process of vascular calcification in chronic kidney disease. Int. J. Biol. Sci. 2016, 12, 1236. [Google Scholar] [CrossRef] [Green Version]
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Choukroun, G.; Kamel, S.; Bennis, Y. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol. Dial. Transplant. 2019, 34, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Jover, E.; Silvente, A.; Marin, F.; Martinez-Gonzalez, J.; Orriols, M.; Martinez, C.M.; Puche, C.M.; Valdés, M.; Rodriguez, C.; Hernández-Romero, D. Inhibition of enzymes involved in collagen cross-linking reduces vascular smooth muscle cell calcification. FASEB J. 2018, 32, 4459–4469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Bi, X.; Liu, Y.; Huang, Y.; Xiong, J.; Xu, X.; Xiao, T.; Yu, Y.; Jiang, W.; Huang, Y.; et al. High phosphate-induced calcification of vascular smooth muscle cells is associated with the TLR4/NF-κb signaling pathway. Kidney Blood Press. Res. 2017, 42, 1205–1215. [Google Scholar] [CrossRef]
- Alesutan, I.; Voelkl, J.; Feger, M.; Kratschmar, D.V.; Castor, T.; Mia, S.; Sacherer, M.; Viereck, R.; Borst, O.; Leibrock, C.; et al. Involvement of vascular aldosterone synthase in phosphate-induced osteogenic transformation of vascular smooth muscle cells. Sci. Rep. 2017, 7, 2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- M’Baya-Moutoula, E.; Louvet, L.; Metzinger-Le Meuth, V.; Massy, Z.A.; Metzinger, L. High inorganic phosphate concentration inhibits osteoclastogenesis by modulating miR-223. Biochim. Biophys. Acta-(Bba)-Mol. Basis Dis. 2015, 1852, 2202–2212. [Google Scholar] [CrossRef] [Green Version]
- Ciceri, P.; Elli, F.; Cappelletti, L.; Tosi, D.; Braidotti, P.; Bulfamante, G.; Cozzolino, M. A new in vitro model to delay high phosphate-induced vascular calcification progression. Mol. Cell. Biochem. 2015, 410, 197–206. [Google Scholar] [CrossRef]
- Guerrero, F.; Montes de Oca, A.; Aguilera-Tejero, E.; Zafra, R.; Rodríguez, M.; López, I. The effect of vitamin D derivatives on vascular calcification associated with inflammation. Nephrol. Dial. Transplant. 2012, 27, 2206–2212. [Google Scholar] [CrossRef] [Green Version]
- Shroff, R.C.; McNair, R.; Skepper, J.N.; Figg, N.; Schurgers, L.J.; Deanfield, J.; Rees, L.; Shanahan, C.M. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J. Am. Soc. Nephrol. 2010, 21, 103–112. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, C.P.; Chung, F.M.; Huang, L.L.; Yu, T.H.; Hung, W.C.; Lu, L.F.; Chen, P.Y.; Luo, C.H.; Lee, K.T.; et al. Uremic retention solute indoxyl sulfate level is associated with prolonged QTc interval in early CKD patients. PLoS ONE 2015, 10, e0119545. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Adrahtas, A.; Kelly, D.J.; Kompa, A.R.; Wang, B.H.; Krum, H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur. Heart J. 2010, 31, 1771–1779. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, B.H.; Kompa, A.R.; Lekawanvijit, S.; Krum, H. Antagonists of organic anion transporters 1 and 3 ameliorate adverse cardiac remodelling induced by uremic toxin indoxyl sulfate. Int. J. Cardiol. 2012, 158, 457–458. [Google Scholar] [CrossRef]
- Lano, G.; Laforêt, M.; Von Kotze, C.; Perrin, J.; Addi, T.; Brunet, P.; Poitevin, S.; Burtey, S.; Dou, L. Aryl hydrocarbon receptor activation and tissue factor induction by fluid shear stress and indoxyl sulfate in endothelial cells. Int. J. Mol. Sci. 2020, 21, 2392. [Google Scholar] [CrossRef] [Green Version]
- Chitalia, V.C.; Shivanna, S.; Martorell, J.; Balcells, M.; Bosch, I.; Kolandaivelu, K.; Edelman, E.R. Uremic serum and solutes increase post–vascular interventional thrombotic risk through altered stability of smooth muscle cell tissue factor. Circulation 2013, 127, 365–376. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Ji, S.; Dong, W.; Qi, Y.; Song, W.; Cui, D.; Shi, J. Indolic uremic solutes enhance procoagulant activity of red blood cells through phosphatidylserine exposure and microparticle release. Toxins 2015, 7, 4390–4403. [Google Scholar] [CrossRef] [Green Version]
- Nagy, E.; Liu, Y.; Prentice, K.J.; Sloop, K.W.; Sanders, P.E.; Batchuluun, B.; Hammond, C.D.; Wheeler, M.B.; Durham, T.B. Synthesis and characterization of urofuranoic acids: In vivo metabolism of 2-(2-Carboxyethyl)-4-methyl-5-propylfuran-3-carboxylic acid (CMPF) and effects on in vitro insulin secretion. J. Med. Chem. 2017, 60, 1860–1875. [Google Scholar] [CrossRef]
- D’Apolito, M.; Du, X.; Zong, H.; Catucci, A.; Maiuri, L.; Trivisano, T.; Pettoello-Mantovani, M.; Campanozzi, A.; Raia, V.; Pessin, J.E.; et al. Urea-induced ROS generation causes insulin resistance in mice with chronic renal failure. J. Clin. Investig. 2010, 120, 203–213. [Google Scholar] [CrossRef]
- Cohen, G.; Glorieux, G.; Thornalley, P.; Schepers, E.; Meert, N.; Jankowski, J.; Jankowski, V.; Argiles, A.; Anderstam, B.; Brunet, P.; et al. Review on uraemic toxins III: Recommendations for handling uraemic retention solutes in vitro—Towards a standardized approach for research on uraemia. Nephrol. Dial. Transplant. 2007, 22, 3381–3390. [Google Scholar] [CrossRef] [Green Version]
- Massy, Z.A.; Drueke, T.B. Role of uremic toxins in vascular disease-the end of nihilism? Kidney Int. 2022, 22, S0085–S2538. [Google Scholar] [CrossRef]
- Hung, S.C.; Kuo, K.L.; Huang, H.L.; Lin, C.C.; Tsai, T.H.; Wang, C.H.; Chen, J.W.; Lin, S.J.; Huang, P.H.; Tarng, D.C. Indoxyl sulfate suppresses endothelial progenitor cell–mediated neovascularization. Kidney Int. 2016, 89, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Kojima, M.; Takayanagi, K.; Taguchi, K.; Kobayashi, T. Trimethylamine-N-oxide specifically impairs endothelium-derived hyperpolarizing factor-type relaxation in rat femoral artery. Biol. Pharm. Bull. 2020, 43, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Chen, Y.; Zhu, Z.; Su, X.; Ni, J.; Du, R.; Zhang, R.; Jin, W. p-Cresyl sulfate promotes the formation of atherosclerotic lesions and induces plaque instability by targeting vascular smooth muscle cells. Front. Med. 2016, 10, 320–329. [Google Scholar] [CrossRef]
- Geng, J.; Yang, C.; Wang, B.; Zhang, X.; Hu, T.; Gu, Y.; Li, J. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed. Pharmacother. 2018, 97, 941–947. [Google Scholar] [CrossRef]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Massy, Z.A.; Ivanovski, O.; Nguyen-Khoa, T.; Angulo, J.; Szumilak, D.; Mothu, N.; Phan, O.; Daudon, M.; Lacour, B.; Drüeke, T.B.; et al. Uremia accelerates both atherosclerosis and arterial calcification in apolipoprotein E knockout mice. J. Am. Soc. Nephrol. 2005, 16, 109–116. [Google Scholar] [CrossRef]
- Kuo, K.L.; Zhao, J.F.; Huang, P.H.; Guo, B.C.; Tarng, D.C.; Lee, T.S. Indoxyl sulfate impairs valsartan-induced neovascularization. Redox Biol. 2020, 30, 101433. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Maré, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E. Indoxyl sulfate and p-cresyl sulfate promote vascular calcification and associate with glucose intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef]
- Muteliefu, G.; Shimizu, H.; Enomoto, A.; Nishijima, F.; Takahashi, M.; Niwa, T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am. J. Physiol.-Cell Physiol. 2012, 303, C126–C134. [Google Scholar] [CrossRef] [Green Version]
- Adijiang, A.; Higuchi, Y.; Nishijima, F.; Shimizu, H.; Niwa, T. Indoxyl sulfate, a uremic toxin, promotes cell senescence in aorta of hypertensive rats. Biochem. Biophys. Res. Commun. 2010, 399, 637–641. [Google Scholar] [CrossRef]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transplant. 2008, 23, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Crouthamel, M.H.; Lau, W.L.; Leaf, E.M.; Chavkin, N.W.; Wallingford, M.C.; Peterson, D.F.; Li, X.; Liu, Y.; Chin, M.T.; Levi, M.; et al. Sodium-dependent phosphate cotransporters and phosphate-induced calcification of vascular smooth muscle cells: Redundant roles for PiT-1 and PiT-2. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2625–2632. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.L.; Linnes, M.; Chu, E.Y.; Foster, B.L.; Bartley, B.A.; Somerman, M.J.; Giachelli, C.M. High phosphate feeding promotes mineral and bone abnormalities in mice with chronic kidney disease. Nephrol. Dial. Transplant. 2013, 28, 62–69. [Google Scholar] [CrossRef] [Green Version]
- El-Abbadi, M.M.; Pai, A.S.; Leaf, E.M.; Yang, H.Y.; Bartley, B.A.; Quan, K.K.; Ingalls, C.M.; Liao, H.W.; Giachelli, C.M. Phosphate feeding induces arterial medial calcification in uremic mice: Role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int. 2009, 75, 1297–1307. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Tokumoto, M.; Tatsumoto, N.; Taniguchi, M.; Noguchi, H.; Nakano, T.; Masutani, K.; Ooboshi, H.; Tsuruya, K.; Kitazono, T. Phosphate overload directly induces systemic inflammation and malnutrition as well as vascular calcification in uremia. Am. J.-Physiol.-Ren. Physiol. 2014, 306, F1418–F1428. [Google Scholar] [CrossRef]
- Graciolli, F.G.; Neves, K.R.; dos Reis, L.M.; Graciolli, R.G.; Noronha, I.L.; Moysés, R.M.; Jorgetti, V. Phosphorus overload and PTH induce aortic expression of Runx2 in experimental uraemia. Nephrol. Dial. Transplant. 2009, 24, 1416–1421. [Google Scholar] [CrossRef]
- Hosaka, N.; Mizobuchi, M.; Ogata, H.; Kumata, C.; Kondo, F.; Koiwa, F.; Kinugasa, E.; Akizawa, T. Elastin degradation accelerates phosphate-induced mineralization of vascular smooth muscle cells. Calcif. Tissue Int. 2009, 85, 523–529. [Google Scholar] [CrossRef]
- Yisireyili, M.; Shimizu, H.; Saito, S.; Enomoto, A.; Nishijima, F.; Niwa, T. Indoxyl sulfate promotes cardiac fibrosis with enhanced oxidative stress in hypertensive rats. Life Sci. 2013, 92, 1180–1185. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Kompa, A.R.; Manabe, M.; Wang, B.H.; Langham, R.G.; Nishijima, F.; Kelly, D.J.; Krum, H. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin, indoxyl sulfate. PLoS ONE 2012, 7, e41281. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.T.; Chen, Y.C.; Hsieh, M.H.; Huang, S.Y.; Kao, Y.H.; Chen, Y.A.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. The uremic toxin indoxyl sulfate increases pulmonary vein and atrial arrhythmogenesis. J. Cardiovasc. Electrophysiol. 2015, 26, 203–210. [Google Scholar] [CrossRef]
- Zhu, H.; Pan, L.; Dai, Y.; Zheng, D.; Cai, S. Role of TLR4/MyD88 Signaling Pathway in the Occurrence and Development of Uremia-Induced Myocardial Hypertrophy and Possible Mechanism. Evid.-Based Complement. Altern. Med. 2021, 2021, 7883643. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Cho, H.J.; Adams-Huet, B.; Paek, J.; Hill, K.; Shelton, J.; Amaral, A.P.; Faul, C.; Taniguchi, M.; et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J. Am. Soc. Nephrol. 2015, 26, 1290–1302. [Google Scholar] [CrossRef] [Green Version]
- Prommer, H.U.; Maurer, J.; von Websky, K.; Freise, C.; Sommer, K.; Nasser, H.; Samapati, R.; Reglin, B.; Guimarães, P.; Pries, A.R.; et al. Chronic kidney disease induces a systemic microangiopathy, tissue hypoxia and dysfunctional angiogenesis. Sci. Rep. 2018, 8, 3075. [Google Scholar] [CrossRef]
- Carmona, A.A.; Rigon, B.G.d.S.; Barroso, M.P.B.S.; Hauser, A.B.; Précoma, D.; Bucharles, S.; Noronha, L.D.; Pécoits-Filho, R. Induction of systemic inflammation and thickening of subepicardiac arteries in an animal model of uremia. Braz. J. Nephrol. 2011, 33, 408–412. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Zhu, J.; Zhu, Z.; Ni, J.; Du, R.; Dai, Y.; Chen, Y.; Wu, Z.; Lu, L.; Zhang, R. p-Cresyl sulfate aggravates cardiac dysfunction associated with chronic kidney disease by enhancing apoptosis of cardiomyocytes. J. Am. Heart Assoc. 2015, 4, e001852. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease–associated thrombosis in mice. Blood J. Am. Soc. Hematol. 2017, 129, 2667–2679. [Google Scholar] [CrossRef] [Green Version]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic solute-aryl hydrocarbon receptor-tissue factor axis associates with thrombosis after vascular injury in humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Koppe, L.; Nyam, E.; Vivot, K.; Fox, J.E.M.; Dai, X.Q.; Nguyen, B.N.; Trudel, D.; Attané, C.; Moullé, V.S.; MacDonald, P.E.; et al. Urea impairs β cell glycolysis and insulin secretion in chronic kidney disease. J. Clin. Investig. 2016, 126, 3598–3612. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, J.; Buendía, P.; Merino, A.; Soriano, S.; Esquivias, E.; Martín-Malo, A.; Aljama, P.; Ramírez, R. Cellular senescence determines endothelial cell damage induced by uremia. Exp. Gerontol. 2013, 48, 766–773. [Google Scholar] [CrossRef]
- Li, T.; Gua, C.; Wu, B.; Chen, Y. Increased circulating trimethylamine N-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochem. Biophys. Res. Commun. 2018, 495, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Alix, P.M.; Croze, M.L.; Chambert, S.; Vanholder, R.; Glorieux, G.; Fouque, D.; Soulage, C.O. p-Cresyl glucuronide is a major metabolite of p-cresol in mouse: In contrast to p-cresyl sulphate, p-cresyl glucuronide fails to promote insulin resistance. Nephrol. Dial. Transplant. 2017, 32, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Pillon, N.J.; Vella, R.E.; Croze, M.L.; Pelletier, C.C.; Chambert, S.; Massy, Z.; Glorieux, G.; Vanholder, R.; Dugenet, Y.; et al. p-Cresyl sulfate promotes insulin resistance associated with CKD. J. Am. Soc. Nephrol. 2013, 24, 88–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, P.C.; Chang, J.C.H.; Lin, C.N.; Lee, C.C.; Chen, Y.T.; Chu, P.H.; Kou, G.; Lu, Y.A.; Yang, C.W.; Chen, Y.C. Serum indoxyl sulfate predicts adverse cardiovascular events in patients with chronic kidney disease. J. Formos. Med. Assoc. 2019, 118, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Konje, V.C.; Rajendiran, T.M.; Bellovich, K.; Gadegbeku, C.A.; Gipson, D.S.; Afshinnia, F.; Mathew, A.V.; The Michigan Kidney Translational Core CPROBE Investigator Group. Tryptophan levels associate with incident cardiovascular disease in chronic kidney disease. Clin. Kidney J. 2021, 14, 1097–1105. [Google Scholar] [CrossRef]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients—a prospective cohort study. Nephrol. Dial. Transplant. 2012, 27, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Liabeuf, S.; Glorieux, G.; Lenglet, A.; Diouf, M.; Schepers, E.; Desjardins, L.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin (EUTox) Work Group. Does p-cresylglucuronide have the same impact on mortality as other protein-bound uremic toxins? PLoS ONE 2013, 8, e67168. [Google Scholar] [CrossRef] [Green Version]
- McGovern, A.P.; de Lusignan, S.; van Vlymen, J.; Liyanage, H.; Tomson, C.R.; Gallagher, H.; Rafiq, M.; Jones, S. Serum phosphate as a risk factor for cardiovascular events in people with and without chronic kidney disease: A large community based cohort study. PLos ONE 2013, 8, e74996. [Google Scholar] [CrossRef] [Green Version]
- Kimata, N.; Albert, J.M.; Akiba, T.; Yamazaki, S.; Kawaguchi, Y.; Fukuhara, S.; Akizawa, T.; Saito, A.; Asano, Y.; Kurokawa, K.; et al. Association of mineral metabolism factors with all-cause and cardiovascular mortality in hemodialysis patients: The Japan dialysis outcomes and practice patterns study. Hemodial. Int. 2007, 11, 340–348. [Google Scholar] [CrossRef]
- Slinin, Y.; Foley, R.N.; Collins, A.J. Calcium, phosphorus, parathyroid hormone, and cardiovascular disease in hemodialysis patients: The USRDS waves 1, 3, and 4 study. J. Am. Soc. Nephrol. 2005, 16, 1788–1793. [Google Scholar] [CrossRef]
- Young, E.W.; Albert, J.M.; Satayathum, S.; Goodkin, D.A.; Pisoni, R.L.; Akiba, T.; Akizawa, T.; Kurokawa, K.; Bommer, J.; Piera, L.; et al. Predictors and consequences of altered mineral metabolism: The Dialysis Outcomes and Practice Patterns Study. Kidney Int. 2005, 67, 1179–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, G.A.; Klassen, P.S.; Lazarus, J.M.; Ofsthun, N.; Lowrie, E.G.; Chertow, G.M. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J. Am. Soc. Nephrol. 2004, 15, 2208–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafi, T.; Powe, N.R.; Meyer, T.W.; Hwang, S.; Hai, X.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Hostetter, T.H. Trimethylamine N-oxide and cardiovascular events in hemodialysis patients. J. Am. Soc. Nephrol. 2017, 28, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Six, I.; Flissi, N.; Lenglet, G.; Louvet, L.; Kamel, S.; Gallet, M.; Massy, Z.A.; Liabeuf, S. Uremic toxins and vascular dysfunction. Toxins 2020, 12, 404. [Google Scholar] [CrossRef]
- Berg, A.H.; Drechsler, C.; Wenger, J.; Buccafusca, R.; Hod, T.; Kalim, S.; Ramma, W.; Parikh, S.M.; Steen, H.; Friedman, D.J.; et al. Carbamylation of serum albumin as a risk factor for mortality in patients with kidney failure. Sci. Transl. Med. 2013, 5, 175ra29. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zelnick, L.R.; Huber, M.P.; Wang, K.; Bansal, N.; Hoofnagle, A.N.; Paranji, R.K.; Heckbert, S.R.; Weiss, N.S.; Go, A.S.; et al. Association Between Kidney Clearance of Secretory Solutes and Cardiovascular Events: The Chronic Renal Insufficiency Cohort (CRIC) Study. Am. J. Kidney Dis. 2021, 78, 226–235.e1. [Google Scholar] [CrossRef]
- Shafi, T.; Sirich, T.L.; Meyer, T.W.; Hostetter, T.H.; Plummer, N.S.; Hwang, S.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Powe, N.R. Results of the HEMO Study suggest that p-cresol sulfate and indoxyl sulfate are not associated with cardiovascular outcomes. Kidney Int. 2017, 92, 1484–1492. [Google Scholar] [CrossRef]
- Luce, M.; Bouchara, A.; Pastural, M.; Granjon, S.; Szelag, J.C.; Laville, M.; Arkouche, W.; Fouque, D.; Soulage, C.O.; Koppe, L. Is 3-Carboxy-4-methyl-5-propyl-2-furanpropionate (CMPF) a Clinically Relevant Uremic Toxin in Haemodialysis Patients? Toxins 2018, 10, 205. [Google Scholar] [CrossRef] [Green Version]
- Menon, V.; Greene, T.; Pereira, A.A.; Wang, X.; Beck, G.J.; Kusek, J.W.; Collins, A.J.; Levey, A.S.; Sarnak, M.J. Relationship of phosphorus and calcium-phosphorus product with mortality in CKD. Am. J. Kidney Dis. 2005, 46, 455–463. [Google Scholar] [CrossRef]
- Kaysen, G.A.; Johansen, K.L.; Chertow, G.M.; Dalrymple, L.S.; Kornak, J.; Grimes, B.; Dwyer, T.; Chassy, A.W.; Fiehn, O. Associations of trimethylamine N-oxide with nutritional and inflammatory biomarkers and cardiovascular outcomes in patients new to dialysis. J. Ren. Nutr. 2015, 25, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.J.; Pan, C.F.; Liu, H.L.; Chuang, C.K.; Jayakumar, T.; Wang, T.J.; Chen, H.H.; Wu, C.J. The role of protein-bound uremic toxins on peripheral artery disease and vascular access failure in patients on hemodialysis. Atherosclerosis 2012, 225, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Shafi, T.; Meyer, T.W.; Hostetter, T.H.; Melamed, M.L.; Parekh, R.S.; Hwang, S.; Banerjee, T.; Coresh, J.; Powe, N.R. Free levels of selected organic solutes and cardiovascular morbidity and mortality in hemodialysis patients: Results from the retained organic solutes and clinical outcomes (ROSCO) investigators. PLoS ONE 2015, 10, e0126048. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Lu, Y.C.; Chiu, C.A.; Yu, T.H.; Hung, W.C.; Wang, C.P.; Lu, L.F.; Chung, F.M.; Lee, Y.J.; Tsai, I.T. Levels of indoxyl sulfate are associated with severity of coronary atherosclerosis. Clin. Investig. Med. 2013, 36, E42–E49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Liu, H.L.; Pan, C.F.; Chuang, C.K.; Jayakumar, T.; Wang, T.J.; Chen, H.H.; Wu, C.J. Indoxyl sulfate predicts cardiovascular disease and renal function deterioration in advanced chronic kidney disease. Arch. Med. Res. 2012, 43, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Taki, K.; Tsuruta, Y.; Niwa, T. Indoxyl sulfate and atherosclerotic risk factors in hemodialysis patients. Am. J. Nephrol. 2007, 27, 30–35. [Google Scholar] [CrossRef]
- Lin, C.J.; Wu, C.J.; Pan, C.F.; Chen, Y.C.; Sun, F.J.; Chen, H.H. Serum protein-bound uraemic toxins and clinical outcomes in haemodialysis patients. Nephrol. Dial. Transplant. 2010, 25, 3693–3700. [Google Scholar] [CrossRef] [Green Version]
- Poesen, R.; Evenepoel, P.; de Loor, H.; Kuypers, D.; Augustijns, P.; Meijers, B. Metabolism, protein binding, and renal clearance of microbiota–derived p-cresol in patients with CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1136–1144. [Google Scholar] [CrossRef] [Green Version]
- Poesen, R.; Claes, K.; Evenepoel, P.; de Loor, H.; Augustijns, P.; Kuypers, D.; Meijers, B. Microbiota-derived phenylacetylglutamine associates with overall mortality and cardiovascular disease in patients with CKD. J. Am. Soc. Nephrol. 2016, 27, 3479–3487. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.P.; Lu, L.F.; Yu, T.H.; Hung, W.C.; Chiu, C.A.; Chung, F.M.; Yeh, L.R.; Chen, H.J.; Lee, Y.J.; Houng, J.Y. Serum levels of total p-cresylsulphate are associated with angiographic coronary atherosclerosis severity in stable angina patients with early stage of renal failure. Atherosclerosis 2010, 211, 579–583. [Google Scholar] [CrossRef]
- Nakamura, S.; Sasaki, O.; Nakahama, H.; Inenaga, T.; Kawano, Y. Clinical characteristics and survival in end-stage renal disease patients with arteriosclerosis obliterans. Am. J. Nephrol. 2002, 22, 422–428. [Google Scholar] [CrossRef]
- Merhi, B.; Shireman, T.; Carpenter, M.A.; Kusek, J.W.; Jacques, P.; Pfeffer, M.; Rao, M.; Foster, M.C.; Kim, S.J.; Pesavento, T.E.; et al. Serum phosphorus and risk of cardiovascular disease, all-cause mortality, or graft failure in kidney transplant recipients: An ancillary study of the FAVORIT trial cohort. Am. J. Kidney Dis. 2017, 70, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddington, H.; Hoefield, R.; Sinha, S.; Chrysochou, C.; Lane, B.; Foley, R.N.; Hegarty, J.; New, J.; O’Donoghue, D.J.; Middleton, R.J.; et al. Serum phosphate and mortality in patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 2251–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kestenbaum, B.; Sampson, J.N.; Rudser, K.D.; Patterson, D.J.; Seliger, S.L.; Young, B.; Sherrard, D.J.; Andress, D.L. Serum phosphate levels and mortality risk among people with chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 520–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum trimethylamine-N-oxide is elevated in CKD and correlates with coronary atherosclerosis burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.B.; Morse, B.L.; Djurdjev, O.; Tang, M.; Muirhead, N.; Barrett, B.; Holmes, D.T.; Madore, F.; Clase, C.M.; Rigatto, C.; et al. Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int. 2016, 89, 1144–1152. [Google Scholar] [CrossRef]
- Melamed, M.L.; Plantinga, L.; Shafi, T.; Parekh, R.; Meyer, T.W.; Hostetter, T.H.; Coresh, J.; Powe, N.R. Retained organic solutes, patient characteristics and all-cause and cardiovascular mortality in hemodialysis: Results from the retained organic solutes and clinical outcomes (ROSCO) investigators. BMC Nephrol. 2013, 14, 2579. [Google Scholar] [CrossRef] [Green Version]
- Chinnappa, S.; Tu, Y.K.; Yeh, Y.C.; Glorieux, G.; Vanholder, R.; Mooney, A. Association between protein-bound uremic toxins and asymptomatic cardiac dysfunction in patients with chronic kidney disease. Toxins 2018, 10, 520. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.S.; Chen, J.; Zou, J.Z.; Zhong, Y.H.; Teng, J.; Ji, J.; Chen, Z.W.; Liu, Z.H.; Shen, B.; Nie, Y.X.; et al. Association of indoxyl sulfate with heart failure among patients on hemodialysis. Clin. J. Am. Soc. Nephrol. 2015, 10, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, S.; Hirashiki, A.; Okumura, T.; Yamada, T.; Okamoto, R.; Shinoda, N.; Takeshita, K.; Kondo, T.; Niwa, T.; Murohara, T. Association between indoxyl sulfate and cardiac dysfunction and prognosis in patients with dilated cardiomyopathy. Circ. J. 2013, 77, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Sato, B.; Yoshikawa, D.; Ishii, H.; Suzuki, S.; Inoue, Y.; Takeshita, K.; Tanaka, M.; Kumagai, S.; Matsumoto, M.; Okumura, S.; et al. Relation of plasma indoxyl sulfate levels and estimated glomerular filtration rate to left ventricular diastolic dysfunction. Am. J. Cardiol. 2013, 111, 712–716. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapolski, T.; Kamińska, A.; Kocki, T.; Wysokiński, A.; Urbanska, E.M. Aortic stiffness—Is kynurenic acid a novel marker? Cross-sectional study in patients with persistent atrial fibrillation. PLoS ONE 2020, 15, e0236413. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Myśliwiec, M.; Pawlak, D. Kynurenine pathway–a new link between endothelial dysfunction and carotid atherosclerosis in chronic kidney disease patients. Adv. Med. Sci. 2010, 55, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.H.; Tang, W.H.; Lu, Y.C.; Wang, C.P.; Hung, W.C.; Wu, C.C.; Tsai, I.T.; Chung, F.M.; Houng, J.Y.; Lan, W.C.; et al. Association between hippuric acid and left ventricular hypertrophy in maintenance hemodialysis patients. Clin. Chim. Acta 2018, 484, 47–51. [Google Scholar] [CrossRef]
- Petchey, W.G.; Hawley, C.M.; Johnson, D.W.; Haluska, B.A.; Watkins, T.W.; Isbel, N.M. Multimodality vascular imaging in CKD: Divergence of risk between measured parameters. Nephrol. Dial. Transplant. 2012, 27, 1004–1012. [Google Scholar] [CrossRef] [Green Version]
- Adeney, K.L.; Siscovick, D.S.; Ix, J.H.; Seliger, S.L.; Shlipak, M.G.; Jenny, N.S.; Kestenbaum, B.R. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J. Am. Soc. Nephrol. 2009, 20, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Ketteler, M.; Bongartz, P.; Westenfeld, R.; Wildberger, J.E.; Mahnken, A.H.; Böhm, R.; Metzger, T.; Wanner, C.; Jahnen-Dechent, W.; Floege, J. Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis: A cross-sectional study. Lancet 2003, 361, 827–833. [Google Scholar] [CrossRef]
- Ix, J.H.; De Boer, I.H.; Peralta, C.A.; Adeney, K.L.; Duprez, D.A.; Jenny, N.S.; Siscovick, D.S.; Kestenbaum, B.R. Serum phosphorus concentrations and arterial stiffness among individuals with normal kidney function to moderate kidney disease in MESA. Clin. J. Am. Soc. Nephrol. 2009, 4, 609–615. [Google Scholar] [CrossRef] [Green Version]
- Drechsler, C.; Kalim, S.; Wenger, J.B.; Suntharalingam, P.; Hod, T.; Thadhani, R.I.; Karumanchi, S.A.; Wanner, C.; Berg, A.H. Protein carbamylation is associated with heart failure and mortality in diabetic patients with end-stage renal disease. Kidney Int. 2015, 87, 1201–1208. [Google Scholar] [CrossRef] [Green Version]
- Glorieux, G.; Vanholder, R.; Van Biesen, W.; Pletinck, A.; Schepers, E.; Neirynck, N.; Speeckaert, M.; De Bacquer, D.; Verbeke, F. Free p-cresyl sulfate shows the highest association with cardiovascular outcome in chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 998–1005. [Google Scholar] [CrossRef]
- Scholz, A.; Plate, K.H.; Reiss, Y. Angiopoietin-2: A multifaceted cytokine that functions in both angiogenesis and inflammation. Ann. N. Y. Acad. Sci. 2015, 1347, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Lai, Y.H.; Kuo, C.H.; Lin, Y.L.; Tsai, J.P.; Hsu, B.G. Association between serum indoxyl sulfate levels and endothelial function in non-dialysis chronic kidney disease. Toxins 2019, 11, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jourde-Chiche, N.; Dou, L.; Sabatier, F.; Calaf, R.; Cerini, C.; Robert, S.; Camoin-Jau, L.; Charpiot, P.; Argiles, A.; Dignat-George, F.; et al. Levels of circulating endothelial progenitor cells are related to uremic toxins and vascular injury in hemodialysis patients. J. Thromb. Haemost. 2009, 7, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Domaniewski, T.; Mysliwiec, M.; Pawlak, D. The kynurenines are associated with oxidative stress, inflammation and the prevalence of cardiovascular disease in patients with end-stage renal disease. Atherosclerosis 2009, 204, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Mysliwiec, M.; Pawlak, D. Haemostatic system, biochemical profiles, kynurenines and the prevalence of cardiovascular disease in peritoneally dialyzed patients. Thromb. Res. 2010, 125, e40–e45. [Google Scholar] [CrossRef]
- Pawlak, K.; Domaniewski, T.; Mysliwiec, M.; Pawlak, D. Kynurenines and oxidative status are independently associated with thrombomodulin and von Willebrand factor levels in patients with end-stage renal disease. Thromb. Res. 2009, 124, 452–457. [Google Scholar] [CrossRef]
- Pawlak, K.; Mysliwiec, M.; Pawlak, D. Hypercoagulability is independently associated with kynurenine pathway activation in dialysed uraemic patients. Thromb. Haemost. 2009, 102, 49–55. [Google Scholar] [CrossRef]
- Wu, C.C.; Hsieh, M.Y.; Hung, S.C.; Kuo, K.L.; Tsai, T.H.; Lai, C.L.; Chen, J.W.; Lin, S.J.; Huang, P.H.; Tarng, D.C. Serum indoxyl sulfate associates with postangioplasty thrombosis of dialysis grafts. J. Am. Soc. Nephrol. 2016, 27, 1254–1264. [Google Scholar] [CrossRef] [Green Version]
- Laville, S.M.; Massy, Z.A.; Kamel, S.; Chillon, J.M.; Choukroun, G.; Liabeuf, S. Intestinal Chelators, Sorbants, and Gut-Derived Uremic Toxins. Toxins 2021, 13, 91. [Google Scholar] [CrossRef]
- Loftus, T.J.; Filiberto, A.C.; Ozrazgat-Baslanti, T.; Gopal, S.; Bihorac, A. Cardiovascular and renal disease in chronic critical illness. J. Clin. Med. 2021, 10, 1601. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Chamieh, C.; Liabeuf, S.; Massy, Z. Uremic Toxins and Cardiovascular Risk in Chronic Kidney Disease: What Have We Learned Recently beyond the Past Findings? Toxins 2022, 14, 280. https://doi.org/10.3390/toxins14040280
El Chamieh C, Liabeuf S, Massy Z. Uremic Toxins and Cardiovascular Risk in Chronic Kidney Disease: What Have We Learned Recently beyond the Past Findings? Toxins. 2022; 14(4):280. https://doi.org/10.3390/toxins14040280
Chicago/Turabian StyleEl Chamieh, Carolla, Sophie Liabeuf, and Ziad Massy. 2022. "Uremic Toxins and Cardiovascular Risk in Chronic Kidney Disease: What Have We Learned Recently beyond the Past Findings?" Toxins 14, no. 4: 280. https://doi.org/10.3390/toxins14040280