Development of an Extraction Method of Aflatoxins and Ochratoxin A from Oral, Gastric and Intestinal Phases of Digested Bread by In Vitro Model

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Fortification Levels of OTA and AFs Analysis
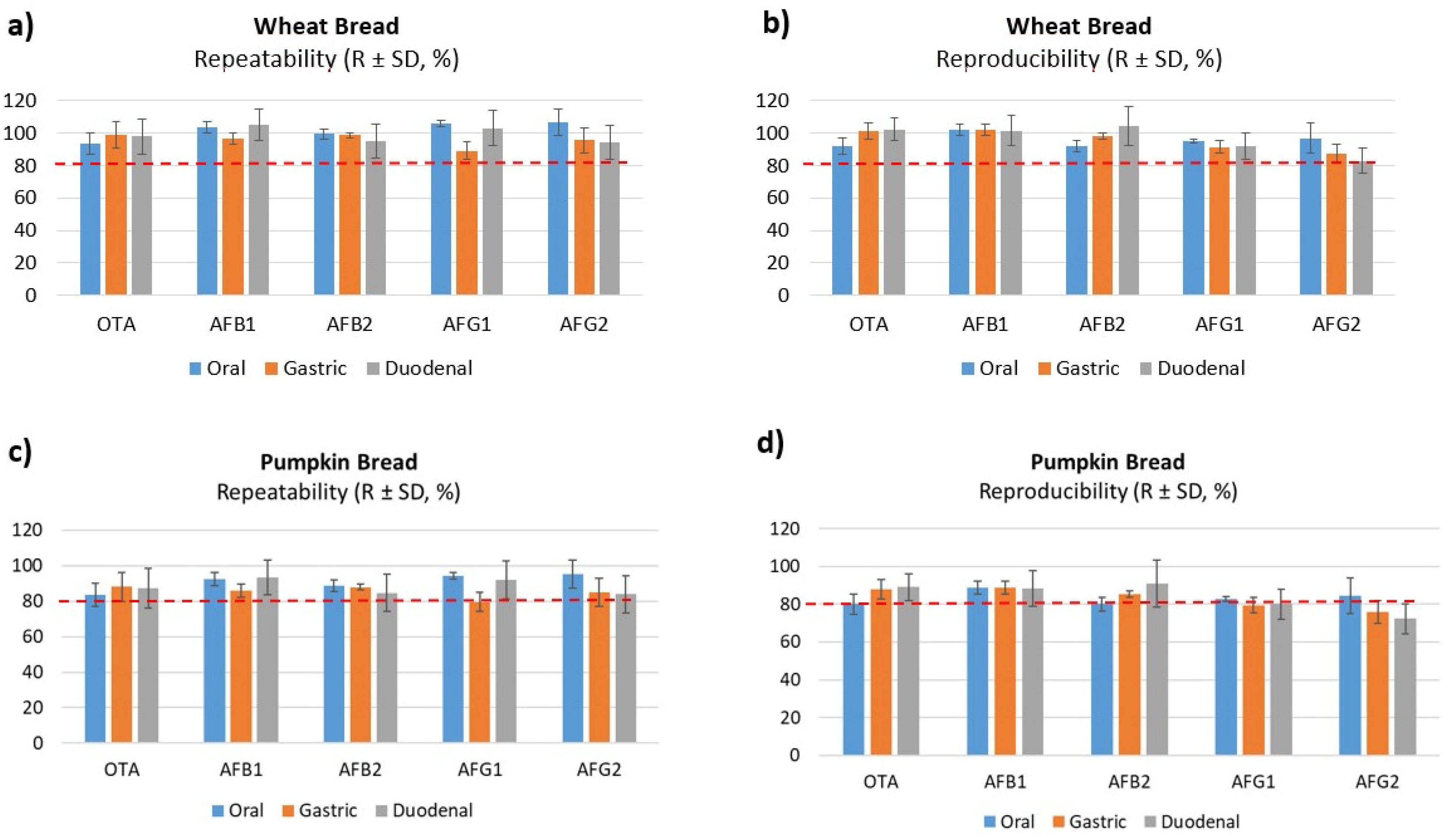
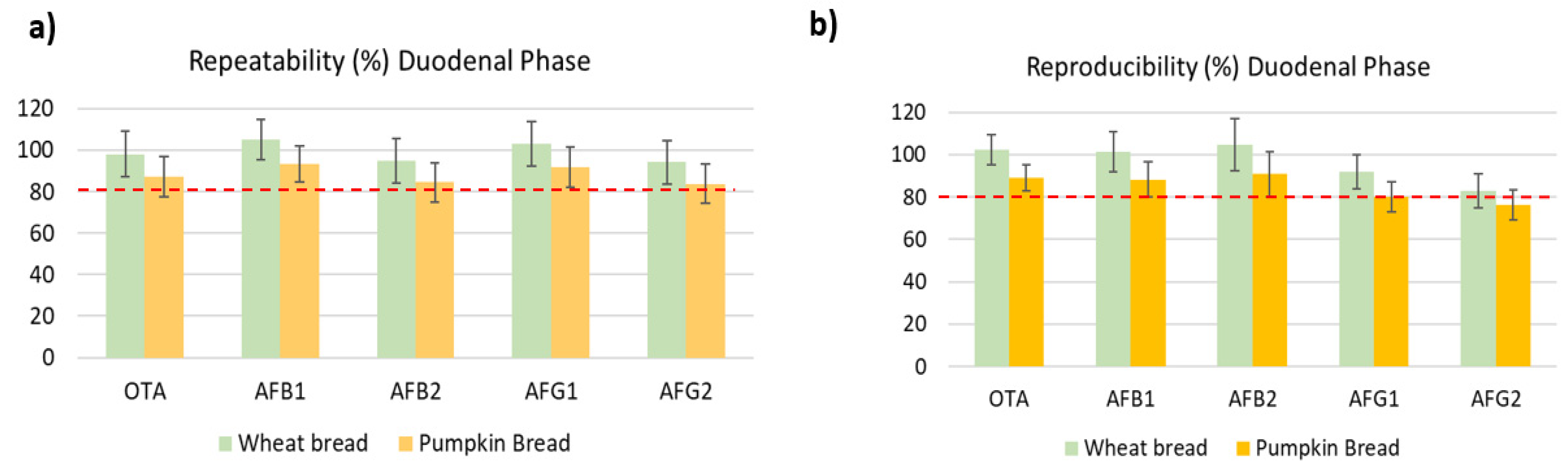
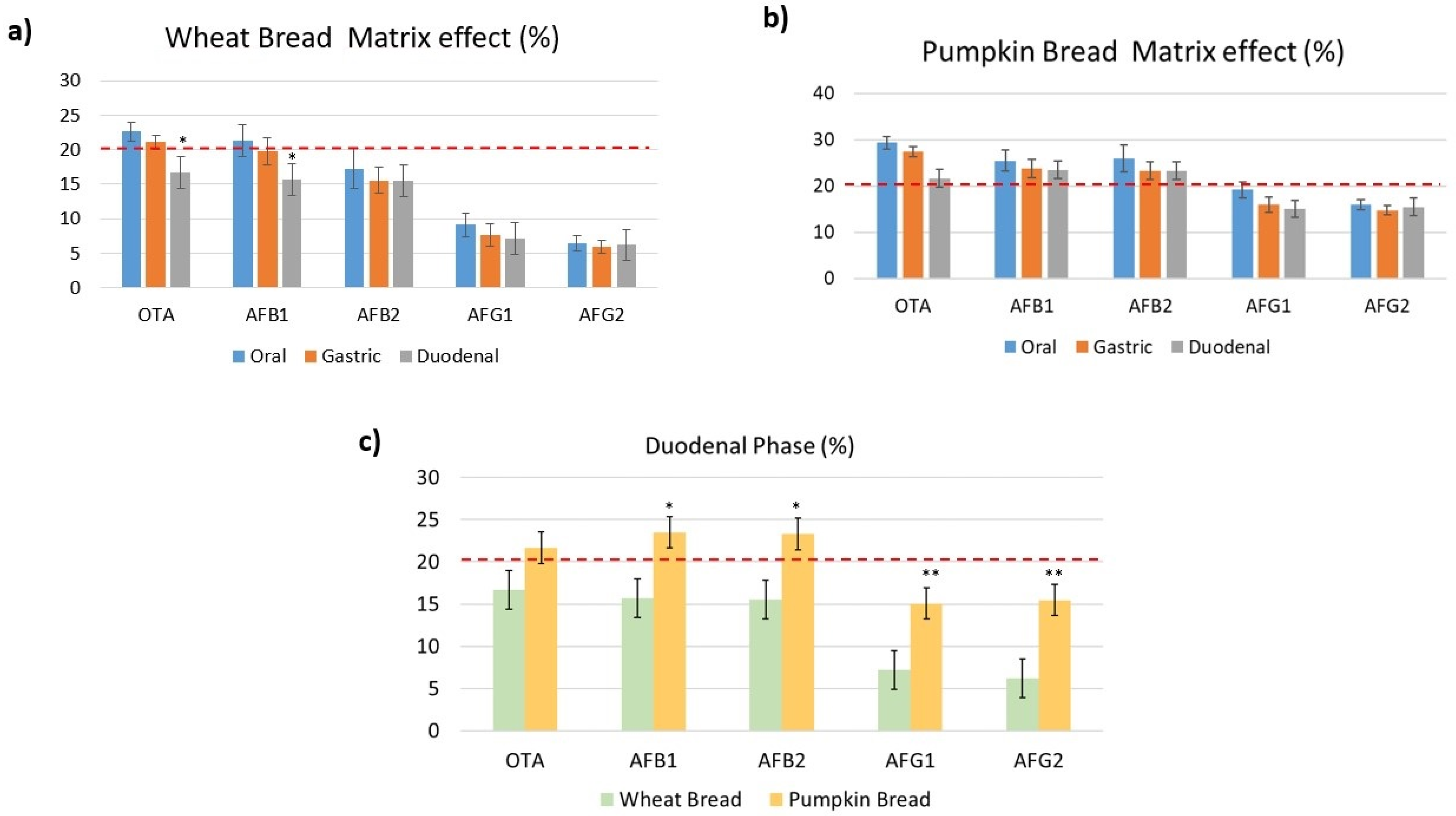
2.2. Validation Results Analysis
2.2.1. Linearity and Sensitivity (LOD and LOQ)

{kind=link}
{kind=link}
{kind=link}
| Bread | Mycotoxin | Oral | Gastric | Duodenal | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LOD (ng/g) | LOQ (ng/g) | Calibration | R2 | LOD (ng/g) | LOQ (ng/g) | Calibration | R2 | LOD (ng/g) | LOQ (ng/g) | Calibration | R2 | ||
| WHEAT BREAD | AFB1 | 0.50 | 1.66 | y = 0.0006x − 2.3155 | 0.9998 | 0.51 | 1.70 | y = 0.0156x − 1.4406 | 0.9999 | 0.48 | 1.60 | y = 0.0006x − 1.0222 | 0.9999 |
| AFB2 | 0.36 | 1.19 | y = 0.0098x + 0.371 | 0.9989 | 0.61 | 2.04 | y = 0.0024x − 31.199 | 0.9961 | 0.34 | 1.14 | y = 0.0095x + 0.407 | 0.9989 | |
| AFG1 | 0.43 | 1.43 | y = 0.001x − 0.3127 | 0.9992 | 0.60 | 2.01 | y = 0.0088x + 0.398 | 0.9989 | 0.41 | 1.38 | y = 0.001x + 0.0547 | 0.9994 | |
| AFG2 | 0.63 | 2.09 | y = 0.0045x − 0.4783 | 0.9997 | 0.91 | 3.04 | y = 0.0021x − 0.4147 | 0.9992 | 0.60 | 2.02 | y = 0.0043x − 0.3119 | 0.9997 | |
| OTA | 0.37 | 1.24 | y = 0.0012x + 0.0101 | 0.9999 | 0.73 | 2.44 | y = 0.0045x − 0.5683 | 0.9997 | 0.36 | 1.20 | y = 0.0011x − 0.0378 | 0.9988 | |
| PUMPKIN BREAD | AFB1 | 0.52 | 1.74 | y = 1595.9x + 3720.1 | 0.9995 | 0.54 | 1.79 | y = 422.1x + 14088 | 0.9996 | 0.47 | 1.58 | y = 1666.7x + 1701.4 | 0.9996 |
| AFB2 | 0.37 | 1.25 | y = 99.215x − 32.625 | 0.9986 | 0.64 | 2.14 | y = 984.96x + 2979.2 | 0.9978 | 0.34 | 1.13 | y = 105.26x − 42.784 | 0.9986 | |
| AFG1 | 0.45 | 1.50 | y = 978.79x + 336.58 | 0.9989 | 0.63 | 2.11 | y = 1275.1x + 63547 | 0.9960 | 0.41 | 1.36 | y = 1000x − 54.626 | 0.9991 | |
| AFG2 | 0.66 | 2.19 | y = 219.2x + 107.6 | 0.9994 | 0.96 | 3.19 | y = 784.6x + 43156 | 0.9989 | 0.60 | 2.00 | y = 232.56x + 72.437 | 0.9994 | |
| OTA | 0.39 | 1.30 | y = 818.18x − 8.2376 | 0.9996 | 0.77 | 2.57 | y = 64.262x + 94.415 | 0.9994 | 0.36 | 1.19 | y = 870.03x + 37.897 | 0.9985 | |
2.2.2. Precision and Trueness
2.2.3. Matrix Effect
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Bread Preparation
4.3. In Vitro Digestion Procedure
4.4. Sample Analysis
4.4.1. Extraction of AFs and OTA from the Simulated Physiological Fluids
4.4.2. LC–MS/MS Analysis
4.5. Method Validation Procedure for Mycotoxins Analysis in Each Digested Phase
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- RASFF. The Rapid Alert System for Food and Feed Annual Report 2020; Publications Office of the European Union: Luxembourg, 2021; ISBN 978-92-76-34377-6/2363-0965. [Google Scholar] [CrossRef]
- Brandon, E.F.A.; Oomen, A.G.; Rompelberg, C.J.M.; Versantvoort, C.H.M.; Van Engelen, J.G.M.; Sips, A.J.A.M. Consumer product in vitro digestion model: Bioaccessibility of contaminants and its application in risk assessment. Regul. Toxicol. Pharmacol. 2006, 44, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Arias, C.A.; Marín, S.; Sanchis, V.; Ramos, A.J. Mycotoxin bioaccesibility/absorption assessment using in vitro digestion models: A review. World Mycotoxin J. 2013, 6, 167–184. [Google Scholar] [CrossRef] [Green Version]
- Andrade, P.D.; Caldas, E.D. Aflatoxins in cereals: Worldwide occurrence and dietary risk assessment. World Mycotoxin J. 2015, 8, 415–431. [Google Scholar] [CrossRef]
- Lee, H.J.; Ryu, D. Worldwide occurrence of mycotoxins in cereals and cereal-derived food products: Public health perspectives of their co-occurrence. J. Agric. Food Chem. 2017, 65, 7034–7051. [Google Scholar] [CrossRef] [PubMed]
- Frenich, G.; Vidal, J.; Romero-Gonzalez, L.M.R.; Aguilera-Luiz, M. Simple and high-throughput method for the multimycotoxin analysis in cereals and related foods by ultra-high performance liquid chromatography/tandem mass spectrometry. Food Chem. 2009, 117, 705–712. [Google Scholar] [CrossRef]
- Malir, F.; Ostry, V.; Novotna, E. Toxicity of the mycotoxin ochratoxin A (OTA) in the light of recent data. Toxin Rev. 2016, 1–15. [Google Scholar]
- International Agency for Research on Cancer (IARC). Monographs Agents classified by the IARC Monographs, 2016, 1–116. Available online: http://monographs.iarc.fr/ENG/Classification/ (accessed on 18 January 2021).
- Pereira, J.A.; Oliveira, I.; Sousa, A.; Ferreira, I.C.F.R.; Bento, A.; Estevinho, L. Bioactive properties and chemical composition of six walnut (Juglans regia L.) cultivars. Food Chem. Toxicol. 2008, 466, 2103–2111. [Google Scholar] [CrossRef]
- AbuZahra, H.M.; Rajendran, P.; Bani Ismail, M. Zerumbone Exhibit Protective Effect against Zearalenone Induced Toxicity via Ameliorating Inflammation and Oxidative Stress Induced Apoptosis. Antioxidants 2021, 10, 1593. [Google Scholar] [CrossRef]
- Rajendran, P.; Alzahrani, A.M.; Priya Veeraraghavan, V.; Ahmed, E.A. Anti-Apoptotic Effect of Flavokawain A on Ochratoxin-A-Induced Endothelial Cell Injury by Attenuation of Oxidative Stress via PI3K/AKT-Mediated Nrf2 Signaling Cascade. Toxins 2021, 13, 745. [Google Scholar] [CrossRef]
- European Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union. 2006, L364, 5–24.
- Kabak, B.; Ozbey, F. Assessment of the bioaccessibility of aflatoxins from various food matrices using an in vitro digestion model, and the efficacy of probiotic bacteria in reducing bioaccessibility. J. Food Compos. Anal. 2012, 27, 21–31. [Google Scholar] [CrossRef]
- Saladino, F.; Posarelli, E.; Luz, C.; Luciano, F.B.; Rodriguez-Estrada, M.T.; Mañes, J.; Meca, G. Influence of probiotic microorganisms on aflatoxins B1 and B2 bioaccessibility evaluated with a simulated gastrointestinal digestion. J. Food Compos. Anal. 2018, 68, 128–132. [Google Scholar] [CrossRef] [Green Version]
- Sambruy, Y.; Ferruzza, S.; Ranaldi, G.; De Angelis, I. Intestinal cell culture models. Cell Biol. Toxicol. 2001, 17, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Versantvoort, C.H.M.; Oomen, A.G.; Van de Kamp, E.; Rompelberg, C.J.M.; Sips, A.J.A.M. Applicability of an in vitro digestion model in assessing the bioaccessibility of mycotoxins from food. Food Chem. Toxicol. 2005, 43, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Rebellato, A.P.; Santos Caramês, E.T.; Lima Pallone, J.A.; Oliveira Rocha, L. Mycotoxin bioaccessibility in baby food through in vitro digestion: An overview focusing on risk assessment. Curr. Opin. Food Sci. 2021, 41, 107–115. [Google Scholar] [CrossRef]
- De Angelis, E.; Monaci, L.; Mackie, A.; Salt, L.; Visconti, A. Reprint of Bioaccessibility of T-2 and HT-2 toxins in mycotoxin contaminated bread models submitted to in vitro human digestion. Innov. Food Sci. Emerg. Technol. 2014, 25, 88–96. [Google Scholar] [CrossRef]
- Massarolo, K.C.; Ferreira, C.F.J.; Collazzo, C.C.; Bianchini, A.; Kupski, L.; Badiale-Furlong, E. Resistant starch and hydrothermal treatment of cornmeal: Factors in aflatoxins and fumonisin B1 reduction and bioaccessibility. Food Control 2020, 114, 107274. [Google Scholar] [CrossRef]
- Massarolo, K.C.; Mendoza, J.R.; Verma, T.; Kupski, L.; Badiale-Furlong, E.; Bianchini, A. Fate of aflatoxins in cornmeal during single-screw extrusion: A bioaccessibility approach. LWT 2021, 138, 110734. [Google Scholar] [CrossRef]
- Morrison, D.M.; Ledoux, D.R.; Chester, L.F.B.; Samuels, C.A.N. Occurrence of aflatoxins in rice and in cassava (Manihot esculenta) products (meal, bread) produced in Guyana. Mycotoxin Res. 2019, 35, 75–81. [Google Scholar] [CrossRef]
- Zinedine, A.; Mañes, J. Occurrence and legislation of mycotoxins in food and feed from Morocco. Food Control 2008, 20, 334–344. [Google Scholar] [CrossRef]
- Paíga, P.; Morais, S.; Oliva-Teles, T.; Correia, M.; Delerue-Matos, C.; Duarte, S.C.; Pena, A.; Lino, C.M. Extraction of ochratoxin A in bread samples by the QuEChERS methodology. Food Chem. 2012, 135, 2522–2528. [Google Scholar] [CrossRef] [Green Version]
- Paíga, P.; Morais, S.; Oliva-Teles, T.; Correia, M.; Delerue-Matos, C.; Sousa, A.M.M.; Gonçalves, M.P.; Duarte, S.C.; Pena, A.; Matos Lino, C. Determination of Ochratoxin A in bread: Evaluation of microwave-assisted extraction using an orthogonal composite design coupled with Response Surface Methodology. Food Bioprocess Technol. 2013, 6, 2466–2477. [Google Scholar] [CrossRef] [Green Version]
- Kulahi, A.; Kabak, B. A preliminary assessment of dietary exposure of ochratoxin A in Central Anatolia Region, Turkey. Mycotoxin Res. 2020, 36, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.Z.; Asi, M.R.; Jinap, S.; Rashid, U. Detection of aflatoxins and zearalenone contamination in wheat derived products. Food Control 2014, 35, 223–226. [Google Scholar] [CrossRef]
- Versantvoort, C.H.M.; Van de Kamp, E.; Rompelberg, C.J.M. Development and Applicability of an In Vitro Digestion Model in Assessing the Bioaccessibility of Contaminants from Food. RIVM Report 320102002/2004. Available online: http://www.rivm.nl/bibliotheek/rapporten/320102002.pdf (accessed on 7 July 2021).
- Kabak, B.; Brandon, E.F.A.; Var, I.; Blokland, M.; Sips, A.J.A.M. Effects of probiotic bacteria on the bioaccessibility of aflatoxin B1 and ochratoxin A using an in vitro digestion model under fed conditions. J. Environ. Sci. Health Part B 2009, 44, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Raiola, A.; Meca, G.; Mañes, J.; Ritieni, A. Bioaccessibility of Deoxynivalenol and its natural co-occurrence with Ochratoxin A and Aflatoxin B1 in Italian commercial pasta. Food Chem. Toxicol. 2012, 50, 280–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Commission. Commission decision 2002/657/EC of 12 August 2002 implementing council directive 96/23/EC concerning the performance of analytical methods and the interpretation of results. Off. J. Eur. Comm. 2002, L221, 8–10. [Google Scholar]
- Meca, G.; Mañes, J.; Font, G.; Ruiz, M.J. Study of the potential toxicity of enniatins A, A1, B, B1 by evaluation of duodenal and colonic bioavailability applying an in vitro method by Caco-2 cells. Toxicon 2012, 59, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Brodkorb, A.; Egger, L.; Alminger, M.; Alvito, P.; Assunção, R.; Ballance, S.; Bohn, T.; Bourlieu-Lacanal, C.; Boutrou, R.; Carrière, F.; et al. INFOGEST static in vitro simulation of gastrointestinal food digestion. Nat. Protoc. 2019, 14, 991–1014. [Google Scholar] [CrossRef]
- El Jai, A.; Juan, C.; Juan-García, A.; Mañes, J.; Zinedine, A. Multi-mycotoxin contamination of green tea infusion and dietary exposure assessment in Moroccan population. Food Res. Int. 2021, 140, 109958. [Google Scholar] [CrossRef]
- Juan, C.; Oueslati, S.; Mañes, J.; Berrada, H. Multimycotoxin determination in Tunisian farm animal feed. J. Food Sci. 2019, 84, 3885–3893. [Google Scholar] [CrossRef] [PubMed]
- Commission Regulation (EC) No 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off. J. Eur. Union. 2006, L70, 12–34.



| Mycotoxin | Repeatability (R ± SD, %) | Reproducibility (R ± SD, %) | ||||
|---|---|---|---|---|---|---|
| L1 | L2 | L3 | L1 | L2 | L3 | |
| AFB1 | 102 ± 1 | 110 ± 4 | 103 ± 9 | 106 ± 4 | 101 ± 4 | 107 ± 9 |
| AFB2 | 92 ± 5 | 105 ± 6 | 108 ± 12 | 96 ± 3 | 90 ± 2 | 94 ± 10 |
| AFG1 | 86 ± 3 | 91 ± 4 | 85 ± 8 | 106 ± 8 | 103 ± 5.3 | 105 ± 11 |
| AFG2 | 97 ± 9 | 87 ± 6 | 83 ± 8 | 107 ± 12 | 94 ± 7.9 | 108 ± 10 |
| OTA | 92 ± 13 | 103 ± 17 | 104 ± 18 | 93 ± 12 | 106 ± 13 | 98 ± 12 |
| Mycotoxin | Rt * (min) | Quantitation Transition | Qualifier Transition | ||||
|---|---|---|---|---|---|---|---|
| Q1 (m/z) | Q3 (m/z) | CE (eV) | Q1 (m/z) | Q3 (m/z) | CE (eV) | ||
| AFB1 | 7.81 | 313 | 241 | 41 | 313 | 284 | 39 |
| AFB2 | 7.69 | 315 | 259 | 39 | 315 | 286 | 33 |
| AFG1 | 7.57 | 329 | 311 | 29 | 329 | 243 | 39 |
| AFG2 | 7.47 | 331 | 245 | 39 | 331 | 313 | 27 |
| OTA | 8.75 | 404 | 102 | 97 | 404 | 239 | 27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llorens, P.; Pietrzak-Fiećko, R.; Moltó, J.C.; Mañes, J.; Juan, C. Development of an Extraction Method of Aflatoxins and Ochratoxin A from Oral, Gastric and Intestinal Phases of Digested Bread by In Vitro Model. Toxins 2022, 14, 38. https://doi.org/10.3390/toxins14010038
Llorens P, Pietrzak-Fiećko R, Moltó JC, Mañes J, Juan C. Development of an Extraction Method of Aflatoxins and Ochratoxin A from Oral, Gastric and Intestinal Phases of Digested Bread by In Vitro Model. Toxins. 2022; 14(1):38. https://doi.org/10.3390/toxins14010038
Chicago/Turabian StyleLlorens, Paula, Renata Pietrzak-Fiećko, Juan Carlos Moltó, Jordi Mañes, and Cristina Juan. 2022. "Development of an Extraction Method of Aflatoxins and Ochratoxin A from Oral, Gastric and Intestinal Phases of Digested Bread by In Vitro Model" Toxins 14, no. 1: 38. https://doi.org/10.3390/toxins14010038