Time Course of Renal Transcriptomics after Subchronic Exposure to Ochratoxin A in Fisher Rats

 , and
, and
Abstract
:1. Introduction
2. Results
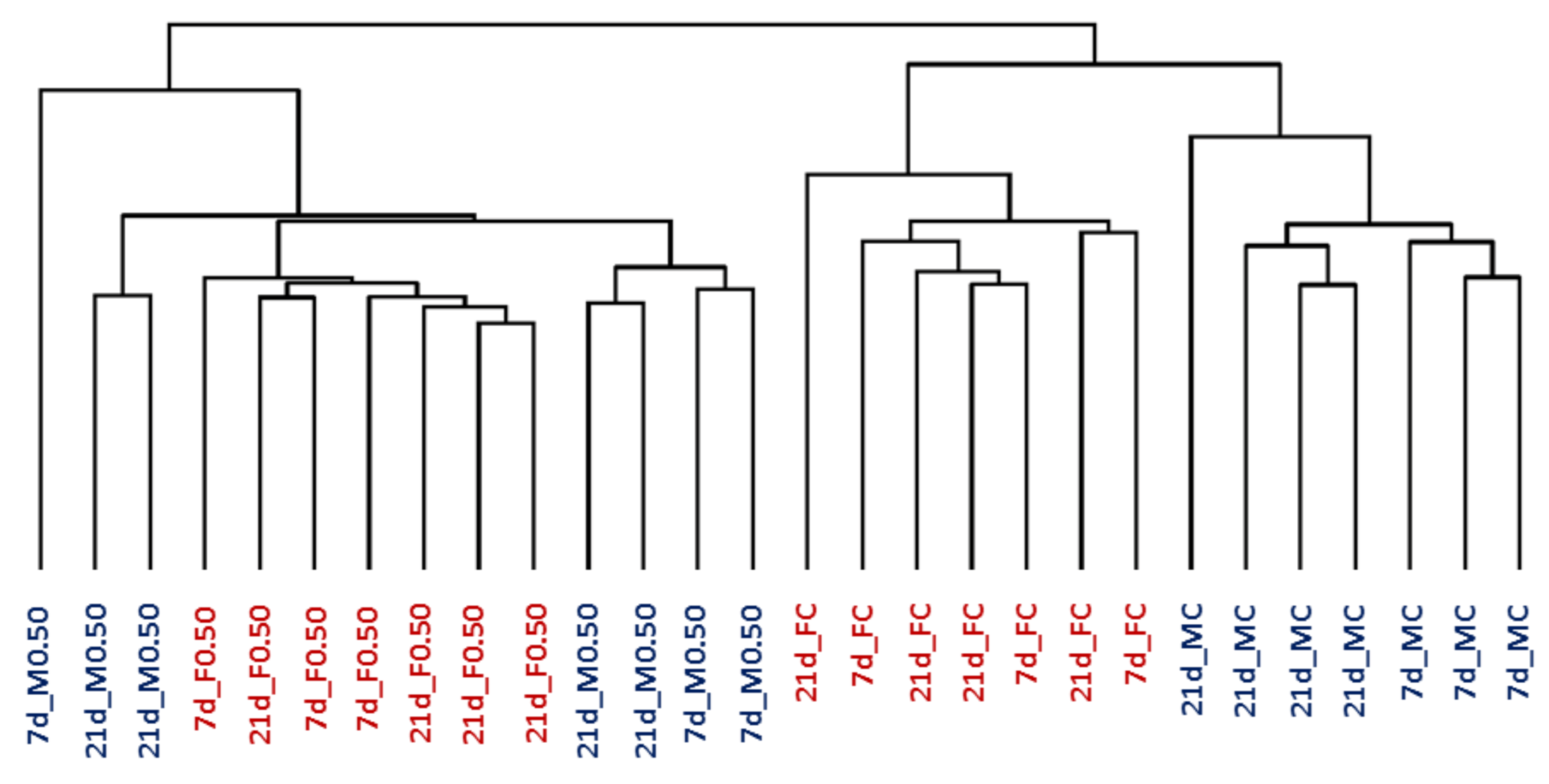
2.1. Gene Expression in Control Rats
2.2. Differentially Expressed Genes after OTA Treatment
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Experimental Design
4.3. RNA Isolation
4.4. Gene Expression Experiments (Microarrays)
4.5. Gene Expression Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Food Safety Authority (EFSA). Contaminants in the food chain on a request from the Commission related to ochratoxin A (OTA) in food. EFSA J. 2006, 365, 1–56. [Google Scholar] [CrossRef]
- Damiano, S.; Longobardi, C.; Andretta, E.; Prisco, F.; Piegari, G.; Squillacioti, C.; Montagnaro, S.; Pagnini, F.; Badino, P.; Florio, S.; et al. Antioxidative Effects of Curcumin on the Hepatotoxicity Induced by Ochratoxin A in Rats. Antioxidants 2021, 10, 125. [Google Scholar] [CrossRef]
- Lock, E.A.; Hard, G.C. Chemically induced renal tubule tumors in the laboratory rat and mouse: Review of the NCI/NTP database and categorization of renal carcinogens based on mechanistic information. Crit. Rev. Toxicol. 2004, 34, 211–299. [Google Scholar] [CrossRef]
- Bendele, A.M.; Carlton, W.W.; Krogh, P.; Lillehoi, E.B. Ochratoxin A carcinogenesis in the (C57BL/6J X C3H) F1 mouse. J. Natl. Cancer Inst. 1985, 75, 733–742. [Google Scholar] [PubMed]
- National Toxicology Program (NTP). Toxicology and carcinogenesis studies of ochratoxin A (CAS No. 303-47-9) in F344/N Rats (Gavage studies). Natl. Toxicol. Program Tech. Rep. Ser. 1989, 358, 1–142. [Google Scholar]
- Castegnaro, M.; Mohr, U.; Pfohl-Leszkowicz, A.; Esteve, J.; Steinmann, J.; Tillmann, T.; Michelon, J.; Bartsch, H. Sex- and strain-specific induction of renal tumors by ochratoxin A in rats correlates with DNA adduction. Int. J. Cancer 1998, 77, 70–75. [Google Scholar] [CrossRef]
- Stoev, S.D. Follow up long term preliminary studies on carcinogenic and toxic effects of ochratoxin A in rats and the putative protection of phenylalanine. Toxicon 2021, 190, 41–49. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research in Cancer (IARC). IARC Monographs on the evaluation of carcinogenic risks to humans. Some naturally occurring substances: Food items and constituents, heterocyclic aromatic amines and mycotoxins. IARC Monogr. 1993, 56, 489–521. [Google Scholar]
- National Toxicology Program (NTP). Report on Carcinogens, 14th ed.; U.S. Department of Health and Human Services, Public Health Service: Research Triangle Park, NC, USA, 2016. [Google Scholar]
- Malir, F.; Ostry, V.; Pfohl-Leszkowicz, A.; Malir, J.; Toman, J. Ochratoxin A: 50 Years of Research. Toxins 2016, 8, 191. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Xie, S.; Xu, F.; Liu, A.; Wang, Y.; Chen, D.; Pan, Y.; Huang, L.; Peng, D.; Wang, X.; et al. Ochratoxin A: Toxicity, oxidative stress and metabolism. Food Chem. Toxicol. 2018, 112, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhang, B.; Dai, Y.; Li, H.; Xu, W. A Review: Epigenetic Mechanism in Ochratoxin A Toxicity Studies. Toxins 2017, 9, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organisation (WHO). Safety evaluation of certain food additives and contaminants. WHO Food Addit. Ser. 2008, 59, 357–429. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as a molecular mechanism of ochratoxin a carcinogenicity. Chem. Res. Toxicol. 2012, 25, 252–262. [Google Scholar] [CrossRef]
- Marin-Kuan, M.; Cavin, C.; Delatour, T.; Schilter, B. Ochratoxin A carcinogenicity involves a complex network of epigenetic mechanisms. Toxicon 2008, 52, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Marin-Kuan, M.; Ehrlich, V.; Delatour, T.; Cavin, C.; Schilter, B. Evidence for a role of oxidative stress in the carcinogenicity of ochratoxin A. J. Toxicol. 2011, 2011, 645361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumora, L.; Zanic-Grubisic, T. A journey through mitogen-activated protein kinase and ochratoxin A interactions. Arh. Hig. Rada. Toksikol. 2009, 60, 449–456. [Google Scholar] [CrossRef] [Green Version]
- Mally, A. Ochratoxin A and mitotic disruption: Mode of action analysis of renal tumor formation by ochratoxin A. Toxicol. Sci. 2012, 127, 315–330. [Google Scholar] [CrossRef]
- Kuiper-Goodman, T.; Hilts, C.; Billiard, S.M.; Kiparissis, Y.; Richard, I.D.; Hayward, S. Health risk assessment of ochratoxin A for all age-sex strata in a market economy. Food Addit. Contam. Part A Chem. Anal. Control Expo Risk Assess. 2010, 27, 212–240. [Google Scholar] [CrossRef] [Green Version]
- Afshari, C.A.; Hamadeh, H.K.; Bushel, P.R. The evolution of bioinformatics in toxicology: Advancing toxicogenomics. Toxicol. Sci. 2011, 120 (Suppl. 1), S225–S237. [Google Scholar] [CrossRef] [Green Version]
- Vettorazzi, A.; van Delft, J.; López de Cerain, A. A review on ochratoxin A transcriptomic studies. Food Chem. Toxicol. 2013, 59, 766–783. [Google Scholar] [CrossRef]
- Marin-Kuan, M.; Nestler, S.; Verguet, C.; Bezencon, C.; Piguet, D.; Mansourian, R.; Holzwarth, J.; Grigorov, M.; Delatour, T.; Mantle, P.; et al. A toxicogenomics approach to identify new plausible epigenetic mechanisms of ochratoxin A carcinogenicity in rat. Toxicol. Sci. 2006, 89, 120–134. [Google Scholar] [CrossRef]
- Arbillaga, L.; Vettorazzi, A.; Gil, A.G.; van Delft, J.H.; García-Jalón, J.A.; López de Cerain, A. Gene expression changes induced by ochratoxin A in renal and hepatic tissues of male F344 rat after oral repeated administration. Toxicol. Appl. Pharmacol. 2008, 230, 197–207. [Google Scholar] [CrossRef]
- Mally, A.; Völkel, W.; Amberg, A.; Kurz, M.; Wanek, P.; Eder, E.; Hard, G.; Dekant, W. Functional, biochemical, and pathological effects of repeated oral administration of ochratoxin A to rats. Chem. Res. Toxicol. 2005, 18, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Kwekel, J.C.; Desai, V.G.; Moland, C.L.; Vijay, V.; Fuscoe, J.C. Sex differences in kidney gene expression during the life cycle of F344 rats. Biol. Sex Differ. 2013, 4, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Penning, T.M. Aldo-keto reductases and bioactivation/detoxication. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 263–292. [Google Scholar] [CrossRef]
- Knight, L.P.; Primiano, T.; Groopman, J.D.; Kensler, T.W.; Sutter, T.R. cDNA cloning, expression and activity of a second human aflatoxin B1-metabolizing member of the aldo-keto reductase superfamily, AKR7A3. Carcinogenesis 1999, 20, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Syvertsen, C.; Størmer, F.C. Oxidation of two hydroxylated ochratoxin A metabolites by alcohol dehydrogenase. Appl. Environ. Microbiol. 1983, 45, 1701–1703. [Google Scholar] [CrossRef] [Green Version]
- Ayed-Boussema, I.; Pascussi, J.M.; Zaied, C.; Maurel, P.; Bacha, H.; Hassen, W. Ochratoxin A induces CYP3A4, 2B6, 3A5, 2C9, 1A1, and CYP1A2 gene expression in primary cultured human hepatocytes: A possible activation of nuclear receptors. Drug Chem. Toxicol. 2012, 35, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Doricakova, A.; Vrzal, R. A food contaminant ochratoxin A suppresses pregnane X receptor (PXR)-mediated CYP3A4 induction in primary cultures of human hepatocytes. Toxicology 2015, 337, 72–78. [Google Scholar] [CrossRef]
- Vettorazzi, A.; Pastor, L.; Guruceaga, E.; López de Cerain, A. Sex-dependent gene expression after ochratoxin A insult in F344 rat kidney. Food Chem. Toxicol. 2019, 123, 337–348. [Google Scholar] [CrossRef] [PubMed]
- El Adlouni, C.; Pinelli, E.; Azémar, B.; Zaoui, D.; Beaune, P.; Pfohl-Leszkowicz, A. Phenobarbital increases DNA adduct and metabolites formed by ochratoxin A; role of CYP2C9 and microsomal glutathione-S-transferase. Environ. Mol. Mutagen. 2000, 35, 123–131. [Google Scholar] [CrossRef]
- Martignoni, M.; Groothuis, G.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Pinelli, E.; Bartsch, H.; Mohr, U.; Castegnaro, M. Sex- and strain-specific expression of cytochrome P450s in ochratoxin A-induced genotoxicity and carcinogenicity in rats. Mol. Carcinog. 1998, 23, 76–85. [Google Scholar] [CrossRef]
- De Groene, E.M.; Hassing, I.G.; Blom, M.J.; Seinen, W.; Fink-Gremmels, J.; Horbach, G.J. Development of human cytochrome P450-expressiong cell lines: Application in mutagenicity testing of ochratoxin A. Cancer Res. 1996, 56, 299–304. [Google Scholar]
- Urbschat, A.; Paulus, P.; von Quernheim, Q.F.; Brück, P.; Badenhoop, K.; Zeuzem, S.; Ramos-Lopez, E. Vitamin D hydroxylases CYP2R1, CYP27B1 and CYP24A1 in renal cell carcinoma. Eur. J. Clin. Investig. 2013, 43, 1282–1290. [Google Scholar] [CrossRef]
- Horváth, H.C.; Lakatos, P.; Kósa, J.P.; Bácsi, K.; Borka, K.; Bises, G.; Nittke, T.; Hershberger, P.A.; Speer, G.; Kállay, E. The candidate oncogene CYP24A1: A potential biomarker for colorectal tumorigenesis. J. Histochem. Cytochem. 2010, 58, 277–285. [Google Scholar] [CrossRef]
- Pastor, L.; Vettorazzi, A.; Enciso, J.M.; González-Peñas, E.; García-Jalón, J.A.; Monreal, J.I.; López de Cerain, A. Sex differences in ochratoxin a toxicity in F344 rats after 7 and 21 days of daily oral administration. Food Chem. Toxicol. 2018, 111, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Limonciel, A.; Jennings, P. A review of the evidence that ochratoxin A is an Nrf2 inhibitor: Implications for nephrotoxicity and renal carcinogenicity. Toxins 2014, 6, 371–379. [Google Scholar] [CrossRef]
- Cavin, C.; Delatour, T.; Marin-Kuan, M.; Holzhäuser, D.; Higgins, L.; Bezencon, C.; Guignard, G.; Junod, S.; Richoz-Pavot, J.; Gremaud, E.; et al. Reduction in antioxidant defenses may contribute to ochratoxin A toxicity and carcinogenicity. Toxicol. Sci. 2007, 96, 30–39. [Google Scholar] [CrossRef]
- Enciso, J.M.; López de Cerain, A.; Pastor, L.; Azqueta, A.; Vettorazzi, A. Is oxidative stress involved in the sex-dependent response to ochratoxin A renal toxicity? Food Chem. Toxicol. 2018, 116, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yu, T.; Qi, X.; Gao, J.; Huang, K.; He, X.; Luo, H.; Xu, W. Limited Link between Oxidative Stress and Ochratoxin A-Induced Renal Injury in an Acute Toxicity Rat Model. Toxins 2016, 8, 373. [Google Scholar] [CrossRef] [Green Version]
- Damiano, S.; Navas, L.; Lombari, P.; Montagnaro, S.; Forte, I.M.; Giordano, A.; Florio, S.; Ciarcia, R. Effects of δ-tocotrienol on ochratoxin A-induced nephrotoxicity in rats. J. Cell. Physiol. 2018, 233, 8731–8739. [Google Scholar] [CrossRef] [PubMed]
- Damiano, S.; Andretta, E.; Longobardi, C.; Prisco, F.; Paciello, O.; Squillacioti, C.; Mirabella, N.; Florio, S.; Ciarcia, R. Effects of Curcumin on the Renal Toxicity Induced by Ochratoxin A in Rats. Antioxidants 2020, 9, 332. [Google Scholar] [CrossRef] [Green Version]
- Damiano, S.; Iovane, V.; Squillacioti, C.; Mirabella, N.; Prisco, F.; Ariano, A.; Amenta, M.; Giordano, A.; Florio, S.; Ciarcia, R. Red orange and lemon extract prevents the renal toxicity induced by ochratoxin A in rats. J. Cell. Physiol. 2020, 235, 5386–5393. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of affymetrix genechip probe level data. Nucleic Acids Res. 2003, 31, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, V.; Carey, S.; Dudoit, R.; Irizarry, R.A.; Huber, W. (Eds.) Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: New York, NY, USA, 2005. [Google Scholar]
- Smyth, G.K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Reference | Species and Strain Dose | Sex | Number of Animals with Tumors/Number of Animals Examined at Each Dose | |
|---|---|---|---|---|
| Bendele et al., 1985 [4] | B6C3F mice 0, 1, 40 ppm | Renal adenoma | Renal carcinoma | |
| M | 0/50, 0/47, 26/50 | 0/50, 0/47, 14/50 | ||
| F | 0/47, 0/45, 0/49 | 0/47, 0/45, 0/49 | ||
| NTP, 1989 [5] | F344 rats 0, 21, 70, 210 µg/kg | Renal adenoma | Renal carcinoma | |
| M | 1/50, 1/51, 6/51, 10/50 | 0/50, 0/51, 16/51, 30/50 | ||
| F | 0/50, 0/51, 1/50, 5/50 | 0/50, 0/51, 1/30, 3/50 | ||
| Castegnaro et al., 1998 [6] | DA and Lewis rats 0, 400 µg/kg | Renal adenoma | Renal carcinoma | |
| M DA | 0/10, 5/20 | 0/10, 20/20 | ||
| F DA | 0/10, 0/40 | 0/10, 0/40 | ||
| M Lewis | 0/10, 0/20 | 0/10, 10/20 | ||
| F Lewis | 0/10, 0/20 | 0/10, 5/20 | ||
| Gene Name | Gene Description | LogFC | |
|---|---|---|---|
| 13 Weeks | 15 Weeks | ||
| Male-biased genes | |||
| Dhrs7 | dehydrogenase/reductase (SDR family) member 7 | 6.85 | 6.97 |
| LOC100364391 | dehydrogenase/reductase (SDR family) member 7-like | 6.43 | 6.24 |
| Slco1a1 | solute carrier organic anion transporter family, member 1a1 | 6.34 | 6.99 |
| Slc22a22 | solute carrier family 22 (organic cation transporter), member 22 | 5.91 | 5.80 |
| Cyp2c11 | cytochrome P450, subfamily 2, polypeptide 11 | 5.18 | 4.92 |
| Eif2s3y | eukaryotic translation initiation factor 2, subunit 3, structural gene Y-linked | 4.12 | 4.25 |
| Slc51b | solute carrier family 51, beta subunit | 4.06 | 3.61 |
| Gc | group specific component | 3.92 | 3.31 |
| Rgn | regucalcin (senescence marker protein-30) | 3.47 | 3.06 |
| Ddx3 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 3 | 3.45 | 3.65 |
| Mybl1 | myeloblastosis oncogene-like 1 | 3.45 | 3.29 |
| LOC102550584 | hornerin-like | 2.85 | 2.89 |
| Sult1b1 | sulfotransferase family, cytosolic, 1B, member 1 | 2.71 | 3.05 |
| Tff3 | trefoil factor 3, intestinal | 2.48 | 1.65 |
| Cyp2d1 | cytochrome P450, family 2, subfamily d, polypeptide 1 | 2.36 | 2.21 |
| Cacng5 | calcium channel, voltage-dependent, gamma subunit 5 | 2.36 | 2.14 |
| Akr1c12l1 | aldo-keto reductase family 1, member C12-like 1 | 2.35 | 2.11 |
| Rarres1 | retinoic acid receptor responder (tazarotene induced) 1 | 2.33 | 1.70 |
| Anxa13 | annexin A13 | 2.23 | 2.41 |
| Melk | maternal embryonic leucine zipper kinase | 2.01 | 1.74 |
| Cyp2d5 | cytochrome P450, family 2, subfamily d, polypeptide 5 | 2.01 | 2.23 |
| Cndp1 | carnosine dipeptidase 1 (metallopeptidase M20 family) | 1.95 | 1.80 |
| Prlr | prolactin receptor | 1.93 | 2.03 |
| LOC500124 | similar to RIKEN cDNA 4921507P07 | 1.92 | 1.70 |
| Gucy1b2 | guanylate cyclase 1, soluble, beta 2 | 1.90 | 1.90 |
| Cyp4a2 | cytochrome P450, family 4, subfamily a, polypeptide 2 | 1.85 | 1.98 |
| Pecr | peroxisomal trans-2-enoyl-CoA reductase | 1.83 | 2.00 |
| Hpgd | hydroxyprostaglandin dehydrogenase 15 (NAD) | 1.81 | 1.73 |
| Mapk10 | mitogen activated protein kinase 10 | 1.80 | 1.84 |
| Akr1c12 | aldo-keto reductase family 1, member C12 | 1.72 | 2.11 |
| Nkd2 | naked cuticle homolog 2 (Drosophila) | 1.67 | 1.50 |
| RGD1564999 | similar to isopentenyl-diphosphate delta isomerase 2 | 2.22 | 2.78 |
| Pzp | pregnancy-zone protein | 1.62 | 2.08 |
| Tmem236 | transmembrane protein 236 | 1.54 | 1.67 |
| Oosp1 | oocyte secreted protein 1 | 1.54 | 1.80 |
| Mlc1 | megalencephalic leukoencephalopathy with subcortical cysts 1 | 1.50 | 1.51 |
| Female-biased genes | |||
| Adh6 | alcohol dehydrogenase 6 (class V) | −6.17 | −5.83 |
| Akr1b7 | aldo-keto reductase family 1, member B7 | −5.25 | −5.72 |
| Ly6al | lymphocyte antigen 6 complex, locus A-like | −3.69 | −4.21 |
| Akr1c2 | aldo-keto reductase family 1, member C2 | −2.80 | −2.96 |
| Slc17a9 | solute carrier family 17 (vesicular nucleotide transporter), member 9 | −2.50 | −2.37 |
| Adh1 | alcohol dehydrogenase 1 (class I) | −2.33 | −1.79 |
| Cmtm2a | CKLF-like MARVEL transmembrane domain containing 2A | −1.83 | −2.17 |
| Col17a1 | collagen, type XVII, alpha 1 | −1.82 | −2.13 |
| Spetex-2H | Spetex-2H protein | −1.77 | −2.55 |
| Slc22a7 | solute carrier family 22 (organic anion transporter), member 7 | −1.73 | −2.44 |
| Col24a1 | collagen, type XXIV, alpha 1 | −1.70 | −2.45 |
| Abcb1b | ATP-binding cassette, subfamily B (MDR/TAP), member 1B | −1.68 | −1.85 |
| Akap17b | A kinase (PRKA) anchor protein 17B | −1.57 | −2.15 |
| Tspan8 | tetraspanin 8 | −1.52 | −1.89 |
| Srsf12 | serine/arginine-rich splicing factor 12 | −1.50 | −1.67 |
| Treatment | DEG | Males | Females |
|---|---|---|---|
| 7 days | Up-regulated (log FC > 0) | 689 | 213 |
| Down-regulated (log FC < 0) | 139 | 281 | |
| Total | 828 | 494 | |
| Log FC > or <1.5 | 19 | 32 | |
| 21 days | Up-regulated (log FC > 0) | 1106 | 1067 |
| Down-regulated (log FC < 0) | 1128 | 1342 | |
| Total | 2234 | 2409 | |
| Log FC > or <1.5 | 136 | 136 |
| Gene Name | Gene Description | LogFC 7 Days | LogFC 21 Days | |
|---|---|---|---|---|
| Females | Akr1b7 | aldo-keto reductase family 1, member B7 | −2.99 | −5.37 |
| Adh6 | alcohol dehydrogenase 6 (class V) | −2.30 | ||
| Akr1c2 | aldo-keto reductase family 1, member C2 | −2.10 | ||
| Slc17a9 | solute carrier family 17, member 9 | −2.12 | ||
| Males | Ucp1 | uncoupling protein 1 (mitochondrial, proton carrier) | −2.72 | |
| Thrsp | thyroid hormone responsive | −2.55 | ||
| Cyp2c11 | cytochrome P450, subfamily 2, polypeptide 11 | −1.96 | −4.43 | |
| Slco1a1 | solute carrier organic anion transporter family, member 1a1 | −5.26 | ||
| Dhrs7 | dehydrogenase/reductase (SDR family) member 7 | −4.49 | ||
| Cyp2d1 | cytochrome P450, family 2, subfamily d, polypeptide 1 | −2.35 | ||
| LOC100364391 | dehydrogenase/reductase (SDR family) member 7-like | −2.27 | ||
| Cyp2d5 | cytochrome P450, family 2, subfamily d, polypeptide 5 | −2.13 | ||
| Slc51b | solute carrier family 51, beta subunit | −2.02 | ||
| Slc22a22 | solute carrier family 22 (organic cation transporter), member 22 | −1.53 | ||
| Cyp24a1 | cytochrome P450, family 24, subfamily a, polypeptide 1 | 1.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastor, L.; Vettorazzi, A.; Guruceaga, E.; López de Cerain, A. Time Course of Renal Transcriptomics after Subchronic Exposure to Ochratoxin A in Fisher Rats. Toxins 2021, 13, 177. https://doi.org/10.3390/toxins13030177
Pastor L, Vettorazzi A, Guruceaga E, López de Cerain A. Time Course of Renal Transcriptomics after Subchronic Exposure to Ochratoxin A in Fisher Rats. Toxins. 2021; 13(3):177. https://doi.org/10.3390/toxins13030177
Chicago/Turabian StylePastor, Laura, Ariane Vettorazzi, Elizabeth Guruceaga, and Adela López de Cerain. 2021. "Time Course of Renal Transcriptomics after Subchronic Exposure to Ochratoxin A in Fisher Rats" Toxins 13, no. 3: 177. https://doi.org/10.3390/toxins13030177