Expansion and Conservation of Biosynthetic Gene Clusters in Pathogenic Pyrenophora spp.

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. PacBio Assembly Statistics for Ptt HRS09122 and HRS09139
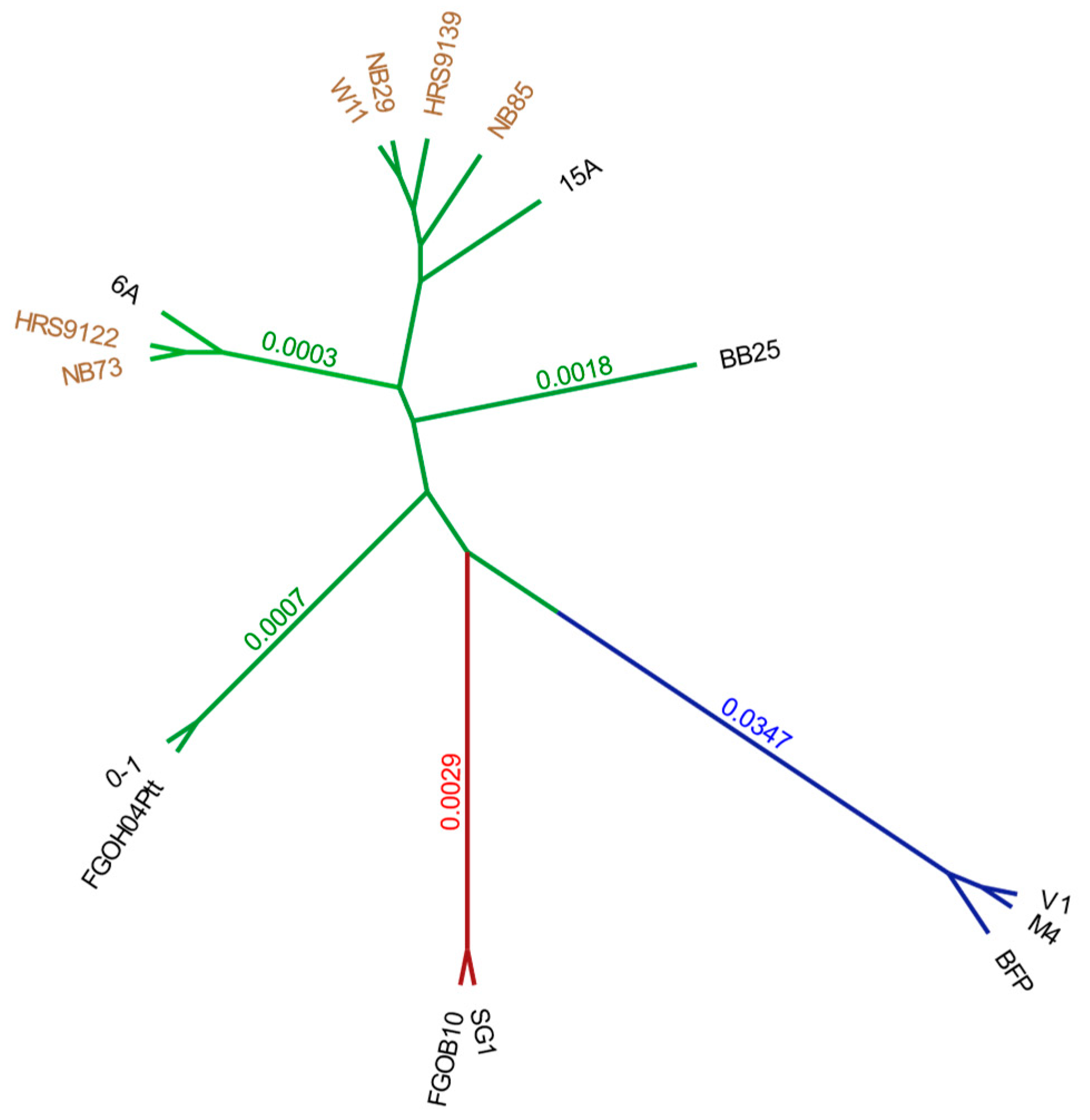
2.2. Genome-Wide Phylogenetic Analysis for Ptt, Ptm and Ptr
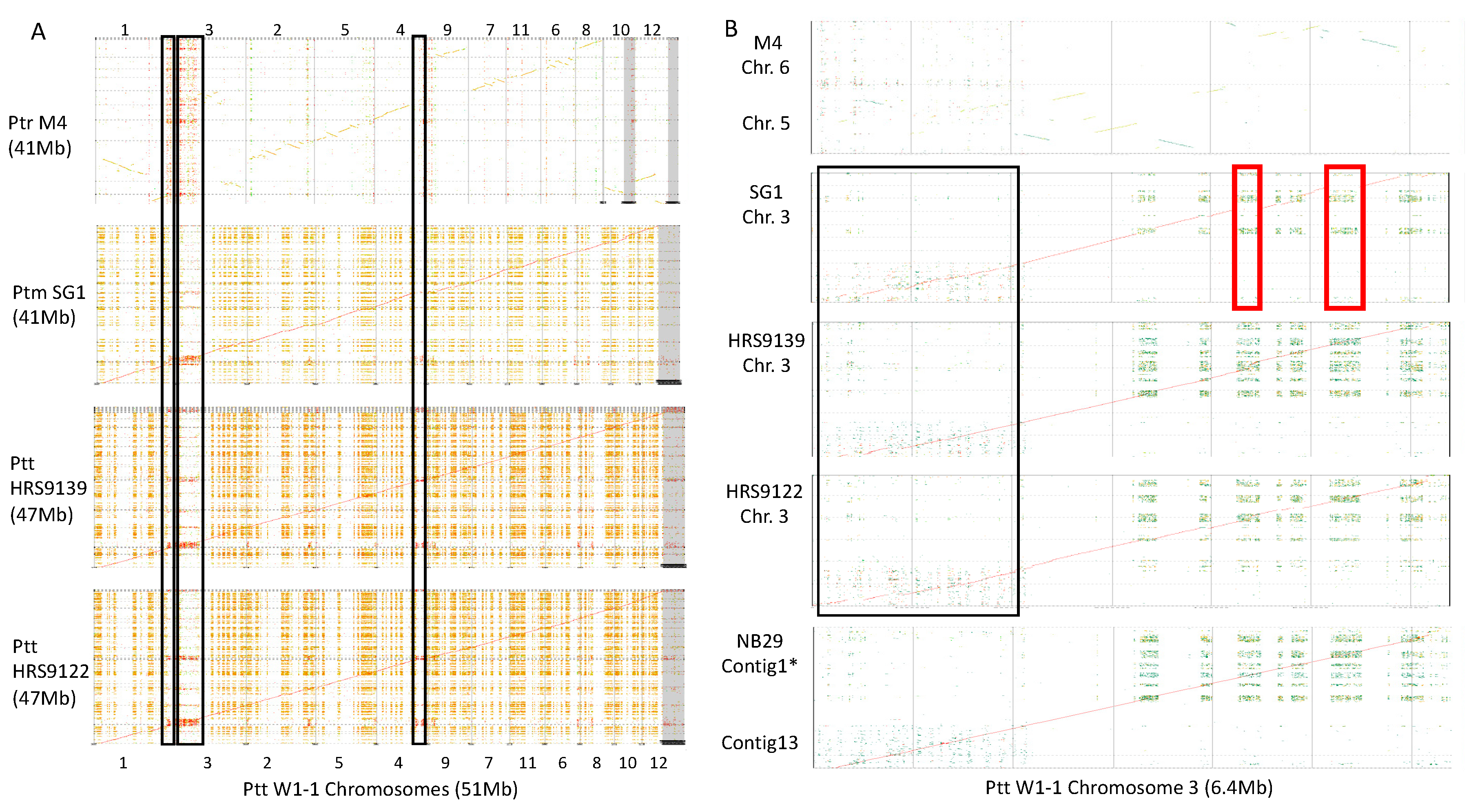
2.3. Genome-Wide Alignments Between Ptt, Ptm and Ptr
2.4. Pyrenophora BGC in Silico Prediction
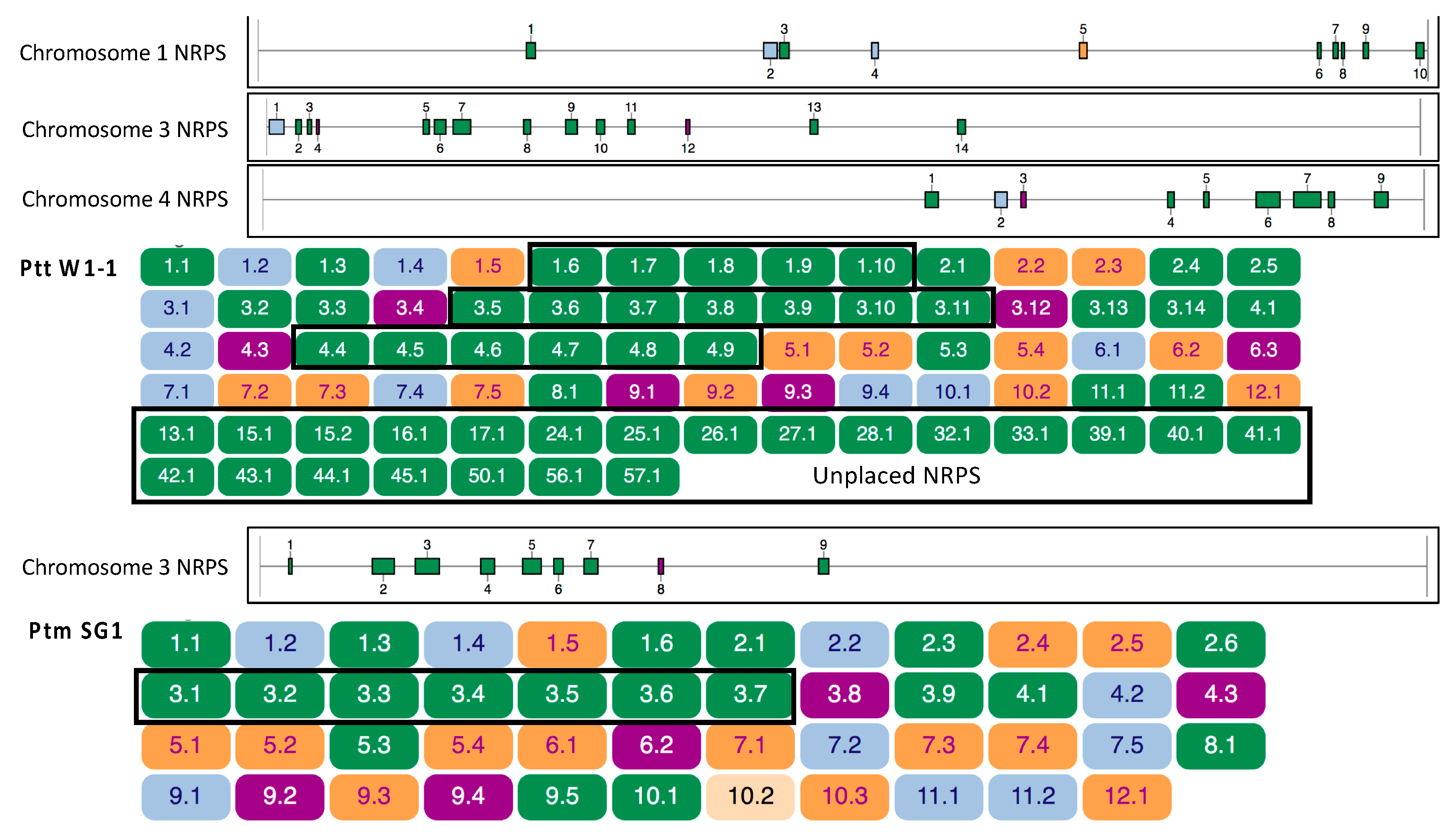
2.5. Orthologous Clustering of NRPS Backbone Protein Sequences
2.6. Orthologous Clustering of T1PKS Backbone Protein Sequences
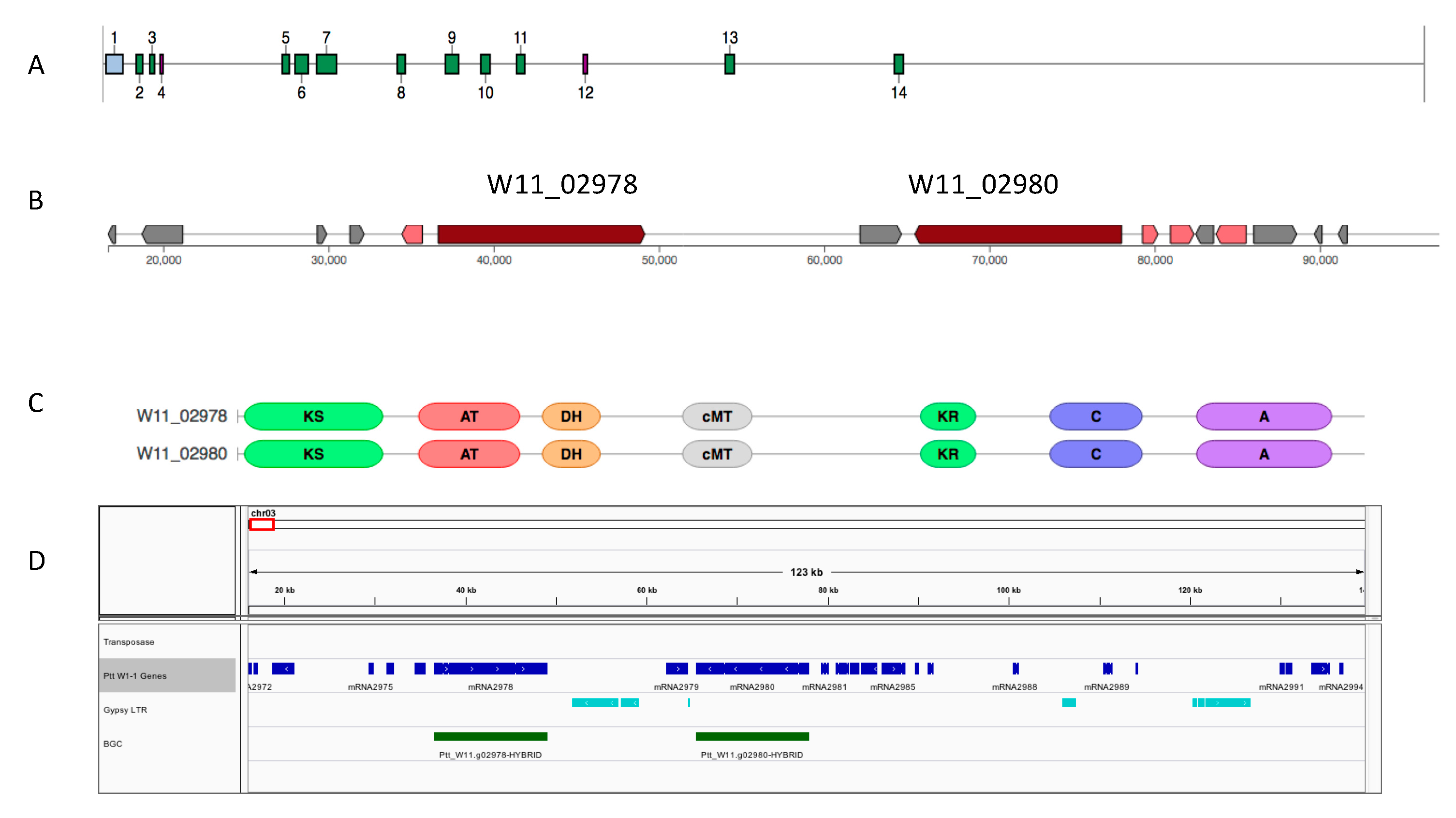
2.7. Biosynthetic Gene Duplications in Pyrenophora
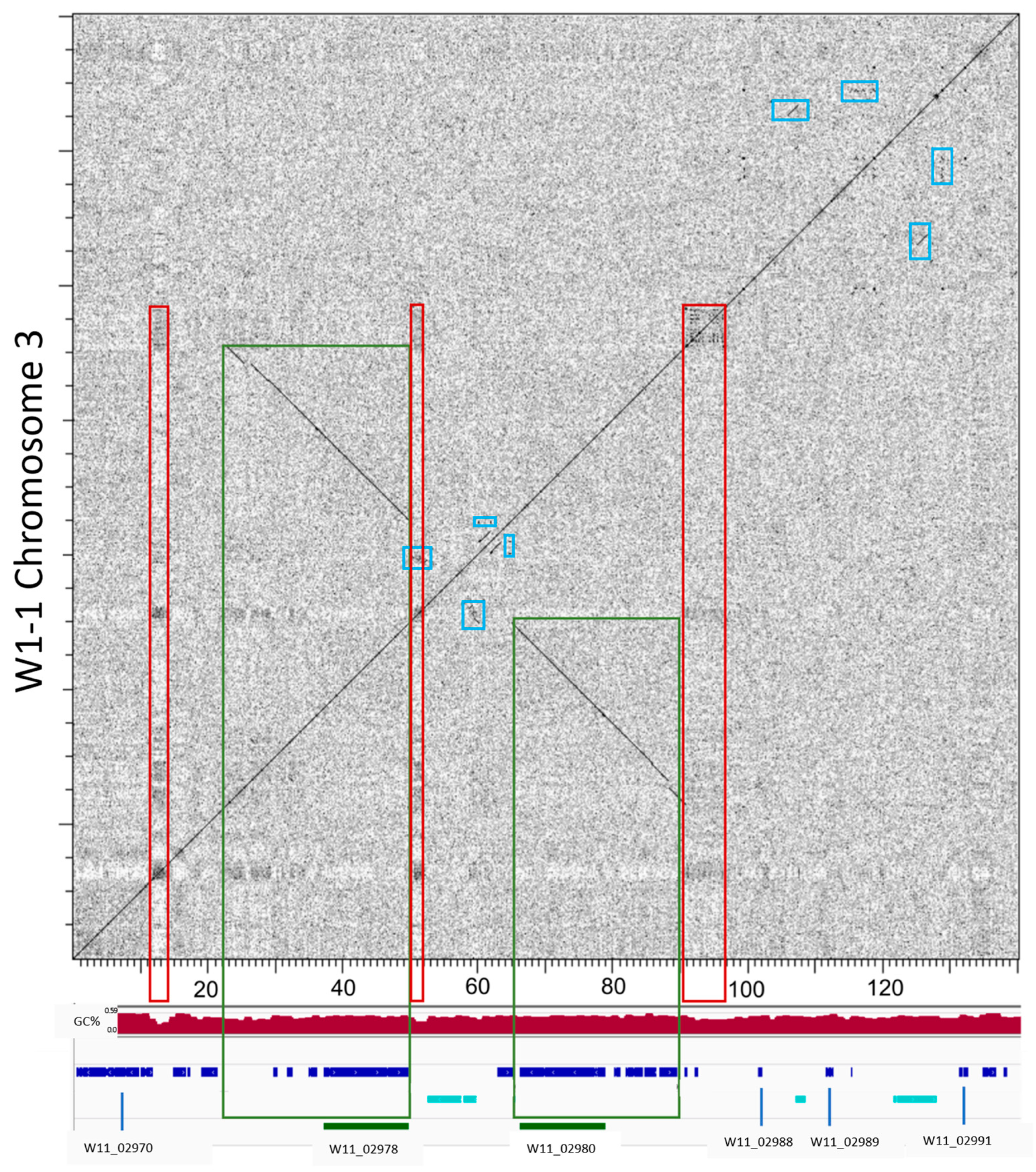
2.8. Pyrenophora teres Regions of NRPS Expansion are Subtelomeric
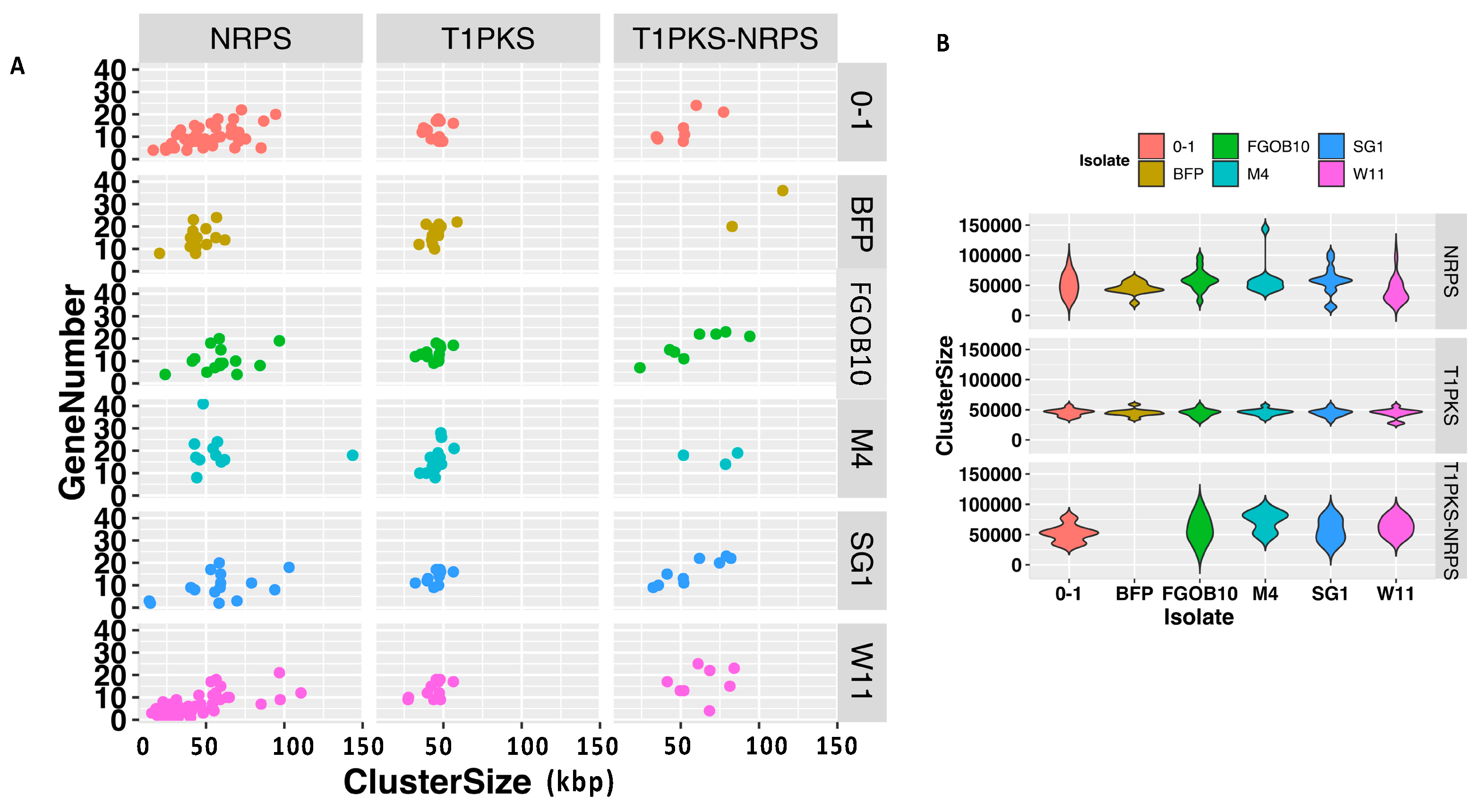
2.9. Comparative Analysis of the Predicted BGCs in Pyrenophora
2.10. Biosynthetic Gene Clusters Conserved in Pyrenophora
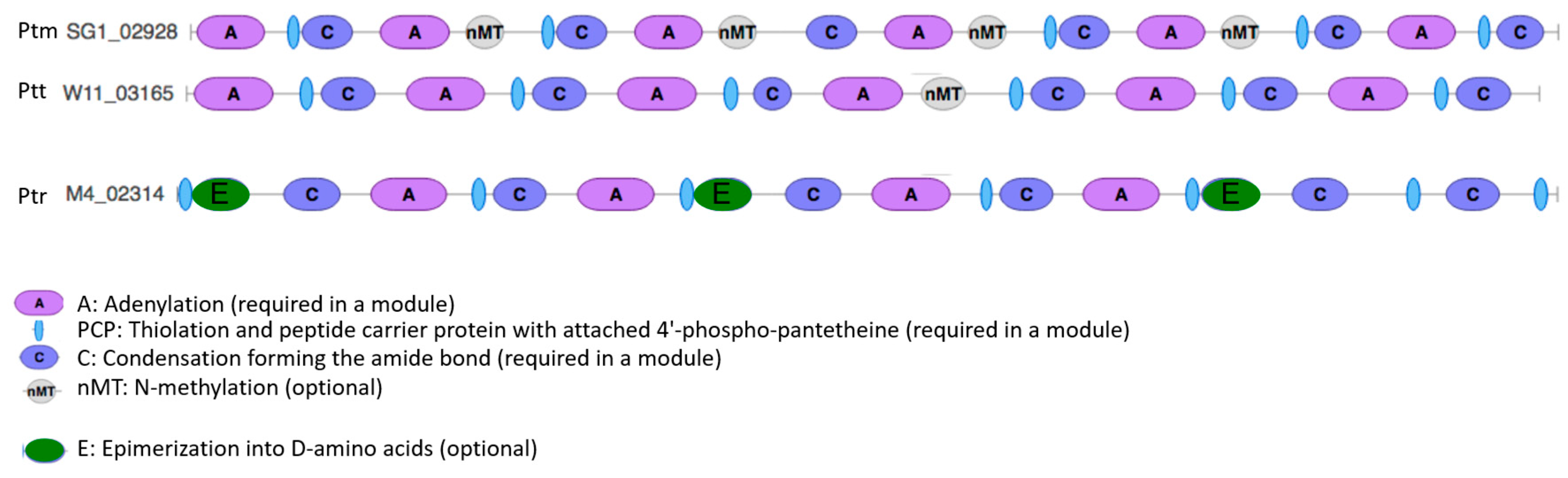
2.11. NRPS Module Domain Nitrogen Methyltransferase Distinguishes P. Teres
3. Discussion
3.1. Pyrenophora Whole Genome Comparative Analysis
3.2. Specialised Metabolites in Pyrenophora
3.3. Absence of Triticone BGC in P. teres Forms
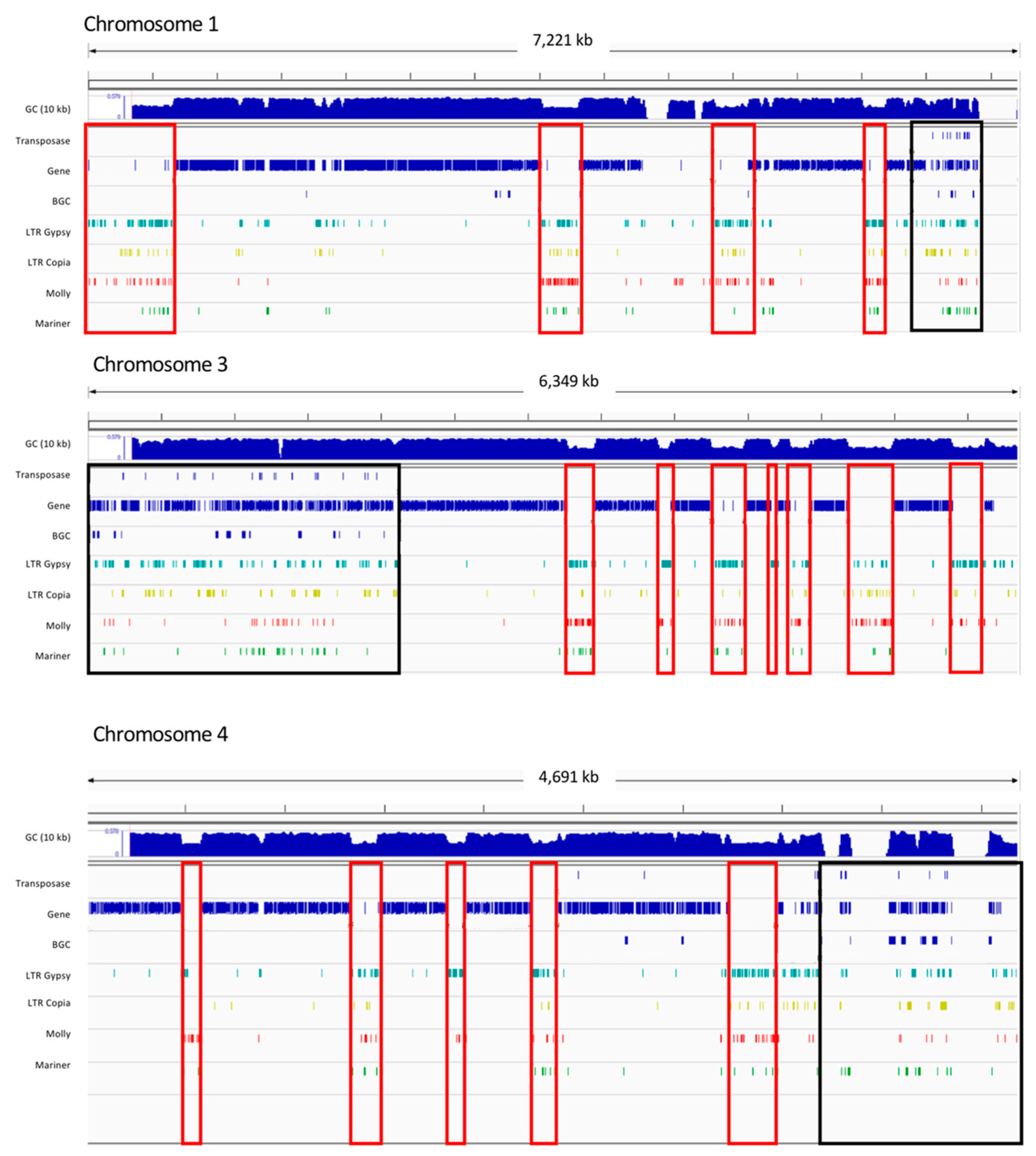
3.4. NRPS Gene Expansion Association with Transposable Elements in P. teres
3.5. Are the Predicted Biosynthetic Gene Clusters in P. teres Complete?
3.6. Pyrenophora Species Have Different Tailoring Enzyme Domains
4. Conclusions
5. Materials and Methods
5.1. Isolates Sequenced in This Study
5.2. DNA Preparation and PacBio Sequencing
5.3. Genome Assemblies
5.4. Specialised Metabolite Gene Cluster Identification
5.5. Other Pyrenophora spp. Genome Sequences Available for Analysis
5.6. In-Silico Search for Syntenic Regions Between Pyrenophora Isolates
5.7. NRPS and PKS Backbone Gene Clustering
5.8. Whole Genome Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schimek, C. Evolution of Special Metabolism in Fungi: Concepts, Mechanisms, and Pathways; Springer: Berlin/Heidelberg, Germany, 2011; pp. 293–329. [Google Scholar]
- Pöggeler, S.; Wöstemeyer, J. (Eds.) Evolution of Fungi and Fungal-Like Organisms; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Inderbitzin, P.; Asvarak, T.; Turgeon, B.G. Six new genes required for production of T-toxin, a polyketide determinant of high virulence of Cochliobolus heterostrophus to maize. Mol. Plant-Microbe Interact. 2010, 23, 458–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osbourn, A. Secondary metabolic gene clusters: Evolutionary toolkits for chemical innovation. Trends Genet. 2010, 26, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeister, D.; Keller, N.P. Natural products of filamentous fungi: Enzymes, genes, and their regulation. Nat. Prod. Rep. 2007, 24, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Hayashi, Y.; Kuzuyama, T.; Furihata, K.; Itoh, N.; Seto, H.; Dairi, T. Biosynthesis of a Natural Polyketide-Isoprenoid Hybrid Compound, Furaquinocin A: Identification and Heterologous Expression of the Gene Cluster. J. Bacteriol. 2006, 188, 1236–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boettger, D.; Hertweck, C. ChemInform Abstract: Molecular Diversity Sculpted by Fungal PKS-NRPS Hybrids. Chemin- 2013, 44, 28–42. [Google Scholar] [CrossRef]
- Khaldi, N.; Seifuddin, F.T.; Turner, G.; Haft, D.; Nierman, W.C.; Wolfe, K.H.; Fedorova, N.D. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Boil. 2010, 47, 736–741. [Google Scholar] [CrossRef] [Green Version]
- Medema, M.H.; Blin, K.; Cimermancic, P.; De Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef]
- Wang, H.; Fewer, D.; Holm, L.; Rouhiainen, L.; Sivonen, K. Atlas of nonribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 9259–9264. [Google Scholar] [CrossRef] [Green Version]
- Ellwood, S.; Syme, R.; Moffat, C.S.; Oliver, R.P. Evolution of three Pyrenophora cereal pathogens: Recent divergence, speciation and evolution of non-coding DNA. Fungal Genet. Boil. 2012, 49, 825–829. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.H.; Friesen, T.L. Identification of Pyrenophora teres f. maculata, Causal Agent of Spot Type Net Blotch of Barley in North Dakota. Plant Dis. 2010, 94, 480. [Google Scholar] [CrossRef]
- Poudel, B.; Martin, A.; Ellwood, S.; Testa, A.C.; McLean, M.; Sutherland, M.W. Rare Pyrenophora teres Hybridization Events Revealed by Development of Sequence-Specific PCR Markers. Phytopathology 2017, 107, 878–884. [Google Scholar] [CrossRef]
- McLean, M.S.; Martin, A.; Gupta, S.; Sutherland, M.W.; Hollaway, G.J.; Platz, G.J. Validation of a new spot form of net blotch differential set and evidence for hybridisation between the spot and net forms of net blotch in Australia. Australas. Plant Pathol. 2014, 43, 223–233. [Google Scholar] [CrossRef]
- Campbell, G.F.; Lucas, J.A.; Braun, U. Evidence of recombination between net- and spot-type populations of Pyrenophora teres as determined by RAPD analysis. Mycol. Res. 2002, 106, 602–608. [Google Scholar] [CrossRef]
- Leišová-Svobodová, L.; Minarikova, V.; Kucera, L.; Ovesná, J.; Minariˇḱova, V. Genetic Diversity of Pyrenophora teres Isolates as Detected by AFLP Analysis. J. Phytopathol. 2005, 153, 569–578. [Google Scholar] [CrossRef]
- Effertz, R.J.; Meinhardt, S.W.; Anderson, J.A.; Jordahl, J.G.; Francl, L. Identification of a Chlorosis-Inducing Toxin from Pyrenophora tritici-repentis and the Chromosomal Location of an Insensitivity Locus in Wheat. Phytopathology 2002, 92, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelakantan, S.; Pocker, A.; Raistrick, H. Studies in the biochemistry of micro-organisms. 98. Tritisporin, an anthraquinone colouring matter of Helminthosporium tritici-vulgaris Nisikado: Structure and synthesis. Biochem. J. 1956, 64, 464–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakuliński, W.; Kachlicki, P.; Sobiczewski, P.; Schollenberger, M.; Zamorski, C.; Łotocka, B.; Sarova, J. Catenarin Production by Isolates of Pyrenophora tritici-repentis (Died.) Drechsler and its Antimicrobial Activity. J. Phytopathol. 2003, 151, 74–79. [Google Scholar] [CrossRef]
- Hallock, Y.F.; Lu, H.S.M.; Clardy, J.; Strobel, G.A.; Sugawara, F.; Samsoedin, R.; Yoshida, S. Triticones, Spirocyclic Lactams from the Fungal Plant Pathogen Drechslera tritici-repentis. J. Nat. Prod. 1993, 56, 747–754. [Google Scholar] [CrossRef]
- Rawlinson, C.; See, P.T.; Moolhuijzen, P.; Li, H.; Moffat, C.S.; Chooi, Y.-H.; Oliver, R.P. The identification and deletion of the polyketide synthase-nonribosomal peptide synthase gene responsible for the production of the phytotoxic triticone A/B in the wheat fungal pathogen Pyrenophora tritici-repentis. Environ. Microbiol. 2019, 21, 4875–4886. [Google Scholar] [CrossRef] [Green Version]
- Weiergang, I.; Jørgensen, H.J.L.; Møller, I.; Friis, P.; Smedegaard-Petersen, V. Correlation between sensitivity of barley to Pyrenophora teres toxins and susceptibility to the fungus. Physiol. Mol. Plant Pathol. 2002, 60, 121–129. [Google Scholar] [CrossRef]
- Nukina, M.; Ikeda, M.; Sassa, T. 2 New Pyrenolides, Fungal Morphogenic Substances Produced by Pyrenophora-Teres (Diedicke) Drechsler. Agric. Biol. Chem. 1980, 44, 2761–2762. [Google Scholar] [CrossRef] [Green Version]
- Nukina, M.; Sassa, T.; Ikeda, M. New Fungal Morphogenic Substance, Pyrenolide a from Pyrenophora-Teres. Tetrahedron Lett. 1980, 21, 301–302. [Google Scholar] [CrossRef]
- Nukina, M.; Hirota, H. Pyrenolide D, a New Cytotoxic Fungal Metabolite fromPyrenophora teres. Biosci. Biotechnol. Biochem. 1992, 56, 1158–1159. [Google Scholar] [CrossRef]
- Coval, S.J.; Hradil, C.M.; Lu, H.S.; Clardy, J.; Satouri, S.; Strobel, G.A. Pyrenoline-a and Pyrenoline-B, 2 New Phytotoxins from Pyrenophora-Teres. Tetrahedron Lett. 1990, 31, 2117–2120. [Google Scholar] [CrossRef]
- Engström, K.; Brishammar, S.; Svensson, C.; Bengtsson, M.; Andersson, R. Anthraquinones from some Drechslera species and Bipolaris sorokiniana. Mycol. Res. 1993, 97, 381–384. [Google Scholar] [CrossRef]
- Manning, V.A.; Pandelova, I.; Dhillon, B.; Wilhelm, L.J.; Goodwin, S.; Berlin, A.M.; Figueroa, M.; Freitag, M.; Hane, J.; Henrissat, B.; et al. Comparative Genomics of a Plant-Pathogenic Fungus, Pyrenophora tritici-repentis, Reveals Transduplication and the Impact of Repeat Elements on Pathogenicity and Population Divergence. G3: Genes|Genomes|Genetics 2013, 3, 41–63. [Google Scholar] [CrossRef] [Green Version]
- Moolhuijzen, P.; See, P.T.; Hane, J.K.; Shi, G.; Liu, Z.; Oliver, R.P.; Moffat, C.S. Comparative genomics of the wheat fungal pathogen Pyrenophora tritici-repentis reveals chromosomal variations and genome plasticity. BMC Genom. 2018, 19, 279. [Google Scholar]
- Syme, R.A.; Martin, A.; Wyatt, N.A.; Lawrence, J.A.; Muria-Gonzalez, M.J.; Friesen, T.L.; Ellwood, S. Transposable Element Genomic Fissuring in Pyrenophora teres Is Associated With Genome Expansion and Dynamics of Host–Pathogen Genetic Interactions. Front. Genet. 2018, 9, 130. [Google Scholar] [CrossRef]
- Wyatt, N.A.; Richards, J.K.; Brueggeman, R.S.; Friesen, T.L. Reference Assembly and Annotation of the Pyrenophora teres f. teres Isolate 0-1. G3: Genes|Genomes|Genetics 2018, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, N.A.; Richards, J.; Brueggeman, R.S.; Friesen, T.L. A Comparative Genomic Analysis of the Barley Pathogen Pyrenophora teres f. teres Identifies Subtelomeric Regions as Drivers of Virulence. Mol. Plant-Microbe Interactions 2020, 33, 173–188. [Google Scholar] [CrossRef]
- Martin, A.; Tao, Y.; McIlroy, J.; Fowler, R.A.; Snyman, L.; Platz, G.J. Genomic Regions Associated with Virulence in Pyrenophora teres f. teres Identified by Genome-Wide Association Analysis and Bi-parental Mapping. Phytopathology 2019. [Google Scholar] [CrossRef]
- Shiryev, S.A.; Papadopoulos, J.S.; Schäffer, A.; Agarwala, R. Improved BLAST searches using longer words for protein seeding. Bioinformatics 2007, 23, 2949–2951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellwood, S.; Piscetek, V.; Mair, W.J.; Lawrence, J.A.; Lopez-Ruiz, F.J.; Rawlinson, C. Genetic variation of Pyrenophora teres f. teres isolates in Western Australia and emergence of a Cyp51A fungicide resistance mutation. Plant Pathol. 2018, 68, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Friesen, T.L.; Stukenbrock, E.H.; Liu, Z.; Meinhardt, S.; Ling, H.; Faris, J.D.; Rasmussen, J.B.; Solomon, P.S.; McDonald, B.A.; Oliver, R.P. Emergence of a new disease as a result of interspecific virulence gene transfer. Nat. Genet. 2006, 38, 953–956. [Google Scholar] [CrossRef]
- McLean, M.S.; Howlett, B.J.; Hollaway, G.J. Epidemiology and control of spot form of net blotch (Pyrenophora teres f. maculata) of barley: A review. Crop. Pasture Sci. 2009, 60, 303. [Google Scholar] [CrossRef]
- Ito, S.; Kuribayashi, K. The ascigerous forms of some graminicolous species of Helminthosporium in Japan. J. Fac. Agric. Hokkaido Imp. Univ. 1931, 29, 85–125. [Google Scholar]
- Barrus, M.F. Yellow-spot disease of wheat in New York state. Plant Dis.Report 1942, 26, 246–247. [Google Scholar]
- Nisikado, Y. Studies on the Helminthosporium diseases of Gramineae in Japan. Ber. Ohara Inst. Landw. Forsch 1929, 4, 111–126. [Google Scholar]
- Shjerve, R.A.; Faris, J.D.; Brueggeman, R.S.; Yan, C.; Zhu, Y.; Koladia, V.; Friesen, T.L. Evaluation of a Pyrenophora teres f. teres mapping population reveals multiple independent interactions with a region of barley chromosome 6H. Fungal Genet. Boil. 2014, 70, 104–112. [Google Scholar] [CrossRef]
- Clare, S.J.; Wyatt, N.A.; Brueggeman, R.S.; Friesen, T.L. Research advances in the Pyrenophora teres-barley interaction. Mol. Plant Pathol. 2019, 21, 272–288. [Google Scholar] [CrossRef] [Green Version]
- Ciuffetti, L.M.; Manning, V.A.; Pandelova, I.; Betts, M.F.; Martinez, J.P. Host-selective toxins, Ptr ToxA and Ptr ToxB, as necrotrophic effectors in the Pyrenophora tritici-repentis-wheat interaction. New Phytol. 2010, 187, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, S.A.; Neupane, A.; Wyatt, N.A.; Richards, J.K.; Faris, J.D.; Xu, S.S.; Brueggeman, R.S.; Friesen, T.L. Characterizing the Pyrenophora teres f. maculata-Barley Interaction Using Pathogen Genetics. G3: Genes|Genomes|Genetics 2017, 7, 2615–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Muñoz, J.C.; Collemare, J. Evolutionary Histories of Type III Polyketide Synthases in Fungi. Front. Microbiol. 2020, 10, 3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Xu, F.; Zeng, J.; Zhan, J. Type III polyketide synthases in natural product biosynthesis. IUBMB Life 2012, 64, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Fujii, I.; Yoshida, N.; Shimomaki, S.; Oikawa, H.; Ebizuka, Y. An Iterative Type I Polyketide Synthase PKSN Catalyzes Synthesis of the Decaketide Alternapyrone with Regio-Specific Octa-Methylation. Chem. Boil. 2005, 12, 1301–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muria-Gonzalez, M.J.; Chooi, Y.-H.; Breen, S.; Solomon, P.S. The past, present and future of secondary metabolite research in the Dothideomycetes. Mol. Plant Pathol. 2014, 16, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Stukenbrock, E.H.; McDonald, B.A. Geographical variation and positive diversifying selection in the host-specific toxin SnToxA. Mol. Plant Pathol. 2007, 8, 321–332. [Google Scholar] [CrossRef]
- McDonald, M.; Ahrén, D.; Simpfendorfer, S.; Milgate, A.; Solomon, P.S. The discovery of the virulence gene ToxA in the wheat and barley pathogen Bipolaris sorokiniana. Mol. Plant Pathol. 2017, 19, 432–439. [Google Scholar] [CrossRef] [Green Version]
- Friesen, T.L.; Holmes, D.J.; Bowden, R.L.; Faris, J.D. ToxA Is Present in the U.S. Bipolaris sorokiniana Population and Is a Significant Virulence Factor on Wheat Harboring Tsn1. Plant Dis. 2018, 102, 2446–2452. [Google Scholar] [CrossRef] [Green Version]
- Cosby, R.L.; Chang, N.-C.; Feschotte, C. Host-transposon interactions: Conflict, cooperation, and cooption. Genome Res. 2019, 33, 1098–1116. [Google Scholar] [CrossRef] [Green Version]
- Hane, J.; Oliver, R.P. RIPCAL: A tool for alignment-based analysis of repeat-induced point mutations in fungal genomic sequences. BMC Bioinform. 2008, 9, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, M.; Taranto, A.P.; Hill, E.; Schwessinger, B.; Liu, Z.; Simpfendorfer, S.; Milgate, A.; Solomon, P.S. Transposon-Mediated Horizontal Transfer of the Host-Specific Virulence Protein ToxA between Three Fungal Wheat Pathogens. mBio 2019, 10, e01515–e01519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.G.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Boil. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Chuma, I.; Hotta, Y.; Tosa, Y. Instability of subtelomeric regions during meiosis in Magnaporthe oryzae. J. Gen. Plant Pathol. 2011, 77, 317–325. [Google Scholar] [CrossRef]
- Ames, R.; Rash, B.M.; Hentges, K.E.; Robertson, D.L.; Delneri, D.; Lovell, S.C. Gene duplication and environmental adaptation within yeast populations. Genome Boil. Evol. 2010, 2, 591–601. [Google Scholar] [CrossRef] [Green Version]
- Ames, R.; Money, D.; Lovell, S.C. Inferring Gene Family Histories in Yeast Identifies Lineage Specific Expansions. PLoS ONE 2014, 9, e99480. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.Z.; Sharma, J.; Gokhale, R.S.; Mohanty, D. In silico analysis of methyltransferase domains involved in biosynthesis of secondary metabolites. BMC Bioinform. 2008, 9, 454. [Google Scholar] [CrossRef] [Green Version]
- Kariyawasam, G.K.; Carter, A.H.; Rasmussen, J.B.; Faris, J.D.; Xu, S.S.; Mergoum, M.; Liu, Z. Genetic relationships between race-nonspecific and race-specific interactions in the wheat–Pyrenophora tritici-repentis pathosystem. Theor. Appl. Genet. 2016, 129, 897–908. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinform. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatic 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Müller, R.; Wohlleben, W.; et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moolhuijzen, P.; See, P.T.; Moffat, C.S. A new PacBio genome sequence of an Australian Pyrenophora tritici-repentis race 1 isolate. BMC Res. Notes 2019, 12, 642–645. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT–the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatic 2011, 27, 1009–1110. [Google Scholar] [CrossRef]
- Emms, D.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Boil. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Boil. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Delcher, A.L.; Salzberg, S.; Phillippy, A.M. Using MUMmer to Identify Similar Regions in Large Sequence Sets. Curr. Protoc. Bioinform. 2003, 10, 10.3.1–10.3.18. [Google Scholar] [CrossRef]
- Haubold, B.; Klötzl, F.; Pfaffelhuber, P. andi: Fast and accurate estimation of evolutionary distances between closely related genomes. Bioinformatic 2014, 31, 1169–1175. [Google Scholar] [CrossRef]
- Retief, J.D. Phylogenetic analysis using PHYLIP. Methods Mol. Boil. (Clifton, N.J.) 2000, 132, 243–258. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | a0-1 | bW1-1 | HRS09122 | HRS09139 |
|---|---|---|---|---|
| Collected | Canada | Western Australia | New South Wales | South Australia |
| Barley cultivar virulence | Harbin | Beecher | Skiff and Gilbert | Fleet |
| Assembly | ||||
| Number contigs/scaffolds* | 55* | 57* | 44 | 91 |
| Total length (Mb) | 46.51 | 53.0 | 47.98 | 50.89 |
| Mean size (Mb) | 845.67 | 931.24 | 109.05 | 559.33 |
| Median size (kb) | 85.05 | 46.63 | 92.01 | 49 |
| Maximum size (Mb) | 5.91 | 7.26 | 5.91 | 6.04 |
| Minimum size (kb) | 27.93 | 18.74 | 21.64 | 1374 |
| Sequences > 10 kb (%) | 55(100.00) | 57(100) | 44(100.00) | 78(85.71) |
| Sequences > 100 kb (%) | 23(41.82) | 12(21.05) | 19(43.18) | 23(25.27) |
| Sequences > 1 Mb (%) | 12(21.82) | 12(21.05) | 15(34.09) | 13(14.29) |
| N50 (Mb) | 4.3 | 4.7 | 3.2 | 3.1 |
| L50 | 5 | 5 | 6 | 6 |
| N80 | 2,252,530 | 3,129,043 | 2,043,292 | 2,506,541 |
| L80 | 10 | 9 | 12 | 11 |
| Genes | ||||
| Protein-coding genes | 11,573 | 11,245 | 10,555 | 10,579 |
| NCBI/ENA BioSample | SAMN07291684 | ERS1459214 | SAMN13065568 | SAMN13065569 |
| NCBI locus tag | NA | PttW1-1 | GD583 | GD582 |
| Isolates | NRPS | T1 PKS | T1 PKS-NRPS Hybrid | NRPS-Like | Terpene | T3PKS | T1 PKS-NRPS-Like | NRPS-Indole | Indole | Total |
|---|---|---|---|---|---|---|---|---|---|---|
| P. triciti-repentis | ||||||||||
| BFP | 13 | 12 | 2 | 4 | 5 | 1 | 0 | 0 | 0 | 37 |
| M4 | 11 | 13 | 3 | 6 | 5 | 1 | 0 | 0 | 0 | 39 |
| V1 | 10 | 14 | 2 | 6 | 5 | 1 | 1 | 0 | 0 | 39 |
| P. teres f. maculata | ||||||||||
| FGOB10 | 15 | 12 | 8 | 3 | 5 | 1 | 0 | 0 | 1 | 45 |
| SG1 | 16 | 12 | 9 | 3 | 5 | 1 | 0 | 0 | 1 | 47 |
| P. teres f. teres | ||||||||||
| 0-1 | 42 | 12 | 7 | 3 | 6 | 1 | 0 | 1 | 0 | 72 |
| 15A | 48 | 11 | 7 | 3 | 5 | 1 | 1 | 1 | 0 | 77 |
| 6A | 51 | 12 | 6 | 3 | 5 | 1 | 0 | 1 | 0 | 79 |
| BB25 | 53 | 12 | 2 | 2 | 4 | 1 | 0 | 1 | 0 | 75 |
| FGOH04 | 43 | 15 | 6 | 4 | 6 | 1 | 0 | 1 | 0 | 76 |
| HRS9122 | 30 | 13 | 7 | 3 | 5 | 1 | 1 | 1 | 0 | 61 |
| HRS9139 | 32 | 13 | 6 | 3 | 5 | 1 | 0 | 1 | 0 | 36 |
| NB29 | 26 | 13 | 6 | 3 | 6 | 1 | 0 | 1 | 0 | 38 |
| NB73 | 34 | 14 | 7 | 3 | 5 | 1 | 0 | 1 | 0 | 39 |
| NB85 | 29 | 13 | 6 | 3 | 6 | 1 | 0 | 1 | 0 | 59 |
| W1-1 | 49 | 12 | 8 | 5 | 6 | 1 | 0 | 1 | 0 | 82 |
| Potential Products | Ptr | Ptm | Ptt |
|---|---|---|---|
| Conserved all pathogens | |||
| Alternapyrone | 1 | 1 | 1 |
| Apicidin | 1 | 1 | 1 |
| Betaenone | 1 | 1 | 1 |
| Dimethylcoprogen | 1 | 1 | 1 |
| Melanin | 1 | 1 | 1 |
| Pestheic acid | 1 | 1 | 1 |
| Conserved P. teres | |||
| ACR-toxin | - | 1 | 1 |
| Azanigerone | - | 1 | 1 |
| Fusarubin | - | 1 | 1 |
| Phomasetin | - | 1 | 1 |
| Not conserved | |||
| Curvupalides/Triticone (Ttc) | 1 | - | - |
| Dehydrocurvularin | 1 | - | - |
| Copalyl diphosphate | - | - | 1 |
| Species | Number of Genomes Searched | Range Percent Identity | Coverage | nMt Domains Genome Count |
|---|---|---|---|---|
| P. tritici-repentis | 4 | 66 | 100 | 1 |
| P. teres f. teres | 12 | 54–100 | 91–100 | 67–93 |
| P. teres f. maculata | 2 | 67–99 | 99–100 | 11–12 |
| P. serminiperda (Illumina) | 1 | 61 | 95 | 1 |
| B. maydis (Illumina) | 2 | 62–72 | 100 | 3 |
| B. cookie (Illumina) | 1 | 64–72 | 100 | 4 |
| B. sorokiniana (Illumina) | 3 | 67–84 | 100 | 6 |
| B. zeicola (Illumina) | 1 | 56–70 | 100 | 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moolhuijzen, P.M.; Muria-Gonzalez, M.J.; Syme, R.; Rawlinson, C.; See, P.T.; Moffat, C.S.; Ellwood, S.R. Expansion and Conservation of Biosynthetic Gene Clusters in Pathogenic Pyrenophora spp. Toxins 2020, 12, 242. https://doi.org/10.3390/toxins12040242
Moolhuijzen PM, Muria-Gonzalez MJ, Syme R, Rawlinson C, See PT, Moffat CS, Ellwood SR. Expansion and Conservation of Biosynthetic Gene Clusters in Pathogenic Pyrenophora spp. Toxins. 2020; 12(4):242. https://doi.org/10.3390/toxins12040242
Chicago/Turabian StyleMoolhuijzen, Paula M., Mariano Jordi Muria-Gonzalez, Robert Syme, Catherine Rawlinson, Pao Theen See, Caroline S. Moffat, and Simon R. Ellwood. 2020. "Expansion and Conservation of Biosynthetic Gene Clusters in Pathogenic Pyrenophora spp." Toxins 12, no. 4: 242. https://doi.org/10.3390/toxins12040242