

Shikonin Binds and Represses PPARγ Activity by Releasing Coactivators and Modulating Histone Methylation Codes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Adipocyte Differentiation
2.3. Oil Red O (ORO) Staining
2.4. Real-Time Quantitative RT-PCR (RT-qPCR)
2.5. RNA-Sequencing and Gene Ontology (GO) Analysis
2.6. Gene Set Enrichment Analysis (GSEA)
2.7. Luciferase Reporter Gene Assays
2.8. Microscale Thermophoresis (MST) Analysis
2.9. Glutathione-S-Transferase (GST) Pull-Down
2.10. ChIP Assays
2.11. Statistical Analysis
3. Results
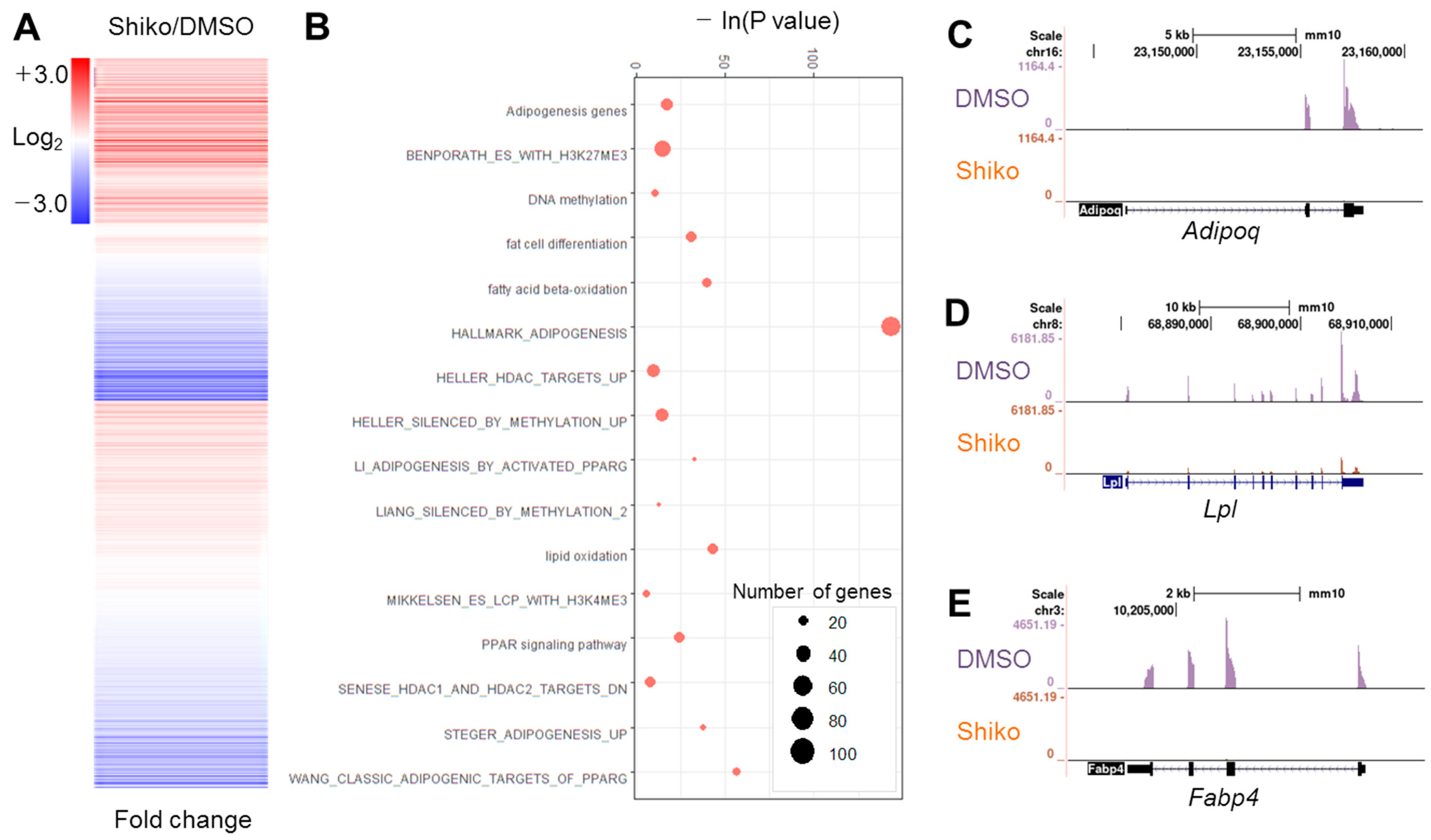
3.1. Shikonin Inhibits Adipogenesis and Downregulates Adipogenic Genes
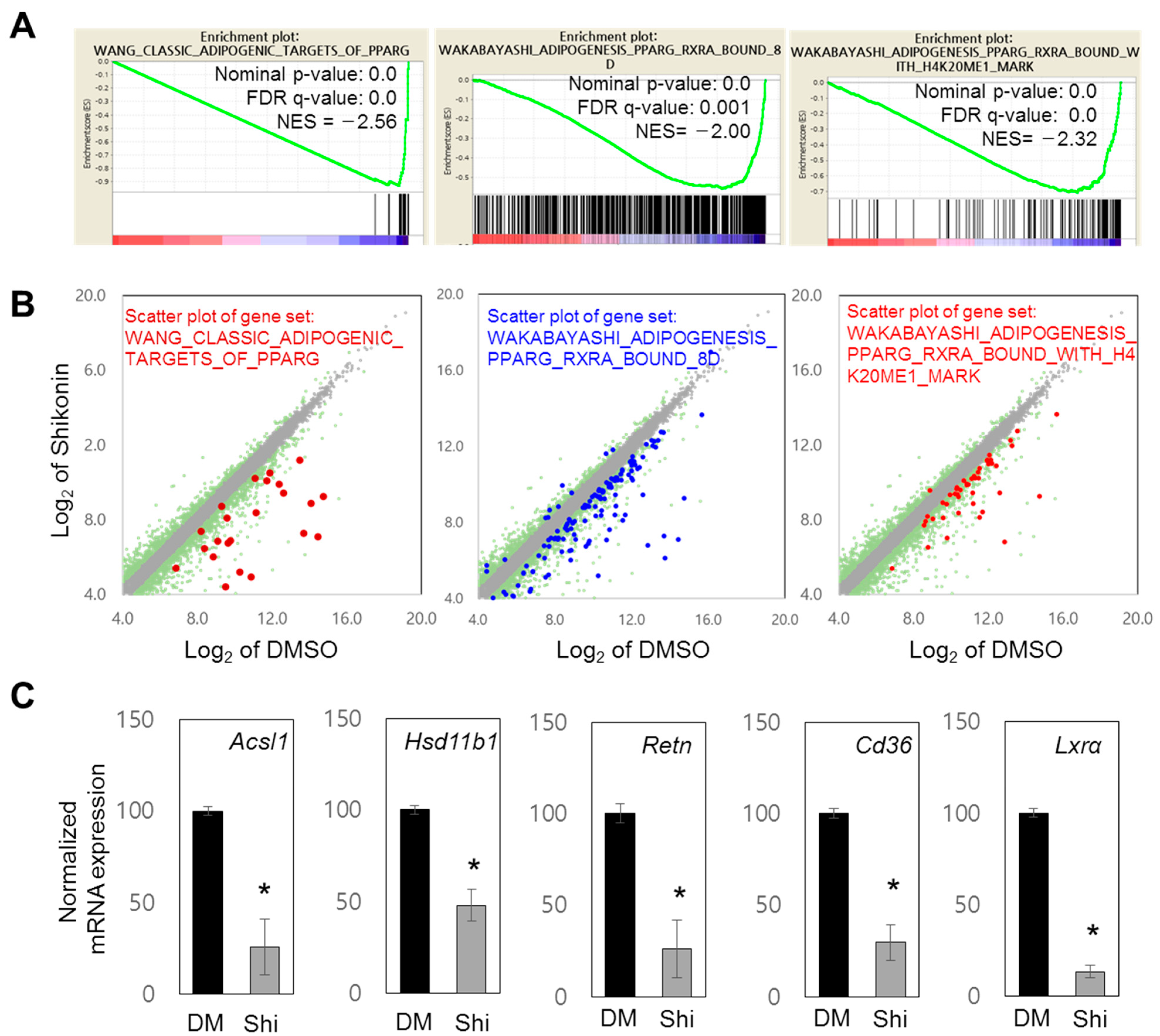
3.2. Shikonin Reduces the Expression of PPARγ Target Genes
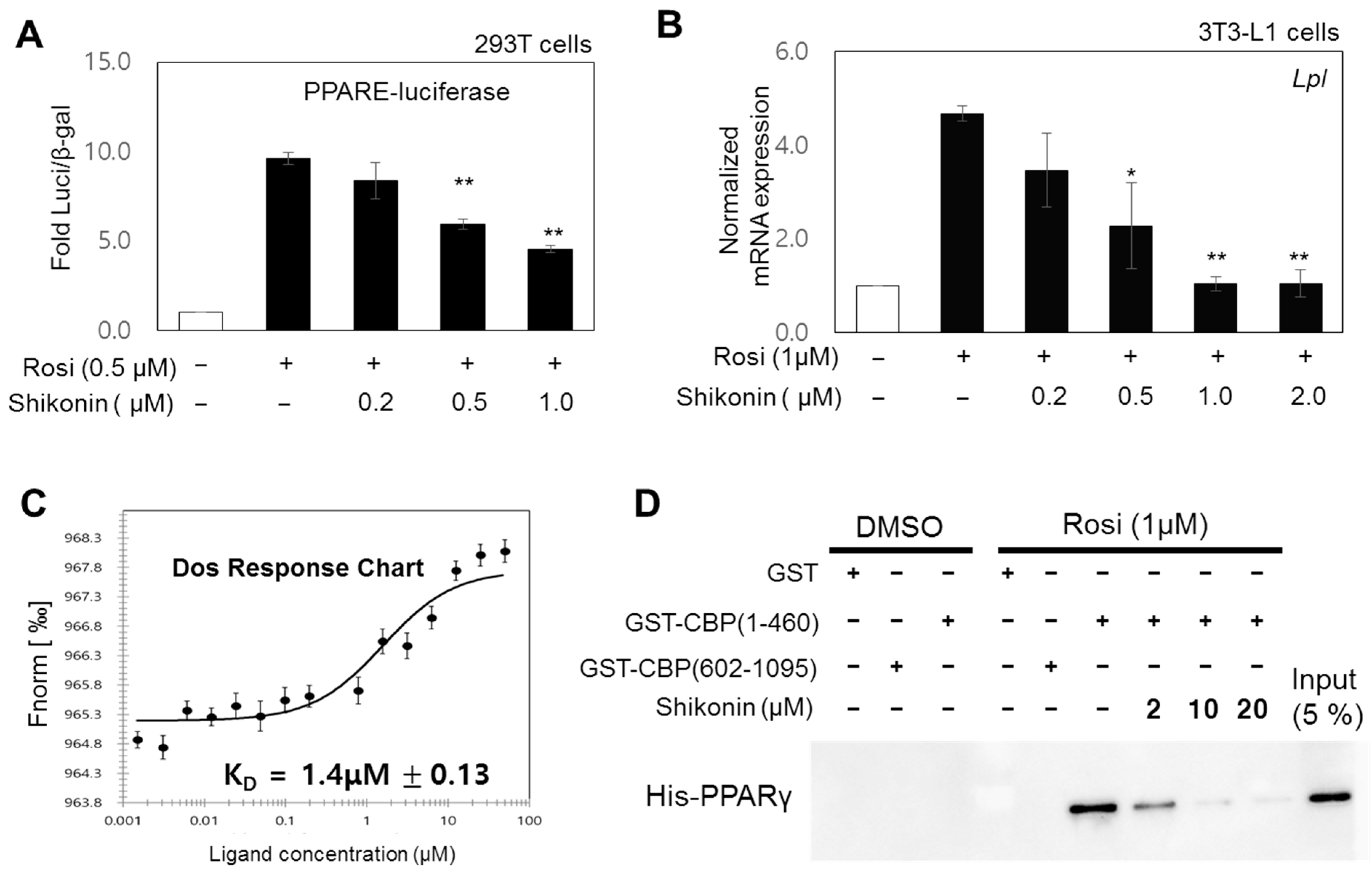
3.3. Shikonin Acts as an Antagonist by Directly Binding PPARγ
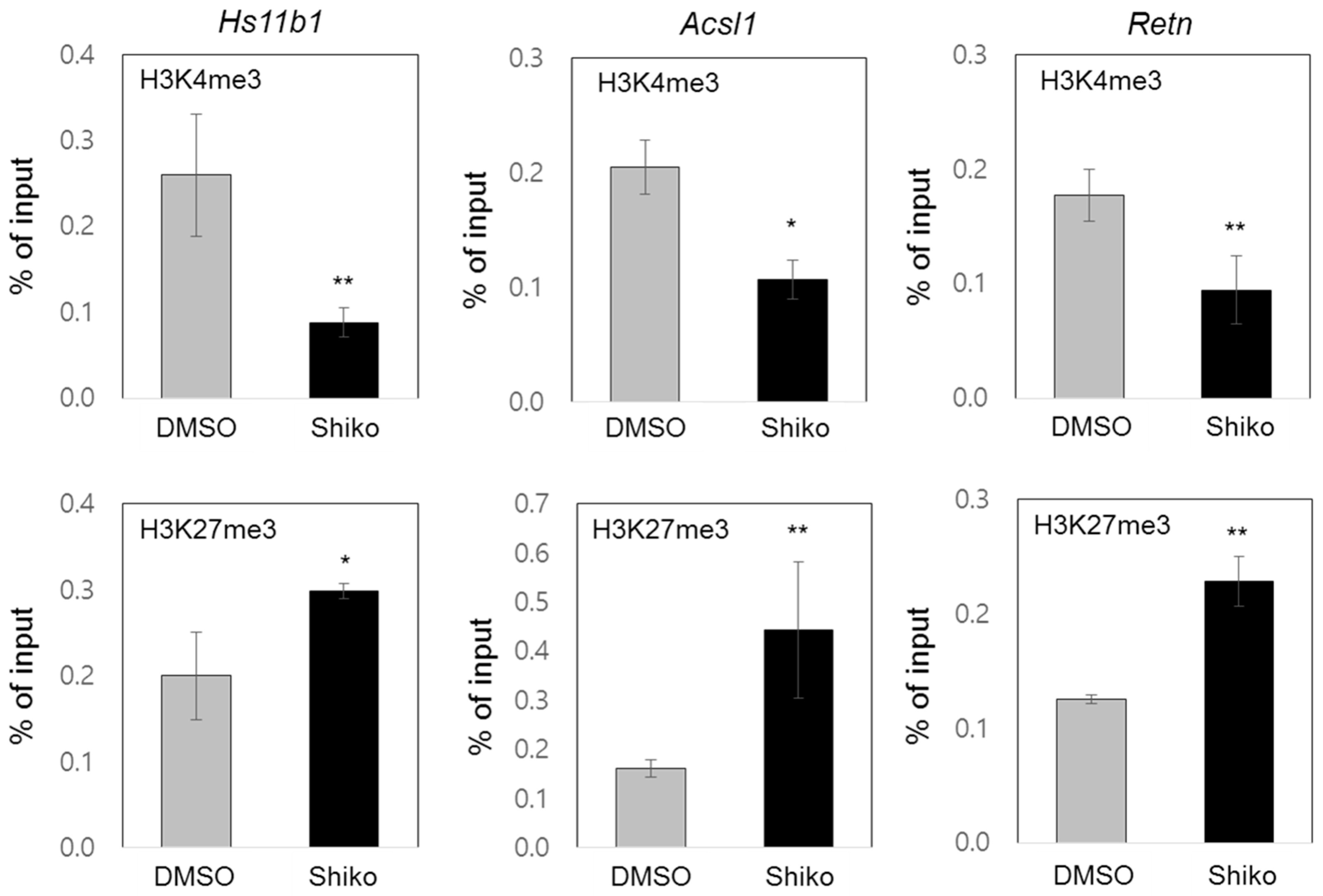
3.4. Shikonin Inhibits PPARγ Target Gene Expression through Enrichment of Active or Repressive Histone Codes on Target Promoters
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. World Obesity Day 2022—Accelerating Action to Stop Obesity. 2022. Available online: https://www.who.int/news/item/04-03-2022-world-obesity-day-2022-accelerating-action-to-stop-obesity (accessed on 1 March 2023).
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T.; et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol. Cell. 1999, 4, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.M.; Gibney, E.R. Epigenetic regulation in obesity. Curr. Opin. Clin. Nutr. 2013, 16, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Isganaitis, E.; Suehiro, H.; Cardona, C. Who’s your daddy? Paternal inheritance of metabolic disease risk. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Herman, H.; Gao, Y.; Lindroth, A.M.; Hu, B.Y.; Murphy, P.J.; Putnam, J.R.; Soloway, P.D. Sequences sufficient for programming imprinted germline DNA methylation defined. PLoS ONE 2012, 7, e33024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, U.H.; Hwang, J.T.; Youn, H.; Kim, E.J.; Um, S.J. Piperine inhibits adipocyte differentiation via dynamic regulation of histone modifications. Phytother. Res. 2019, 33, 2429–2439. [Google Scholar] [CrossRef]
- Park, U.H.; Hwang, J.T.; Youn, H.; Kim, E.J.; Um, S.J. Kaempferol antagonizes adipogenesis by repressing histone H3K4 methylation at PPARgamma target genes. Biochem. Biophys. Res. Commun. 2022, 617, 48–54. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.Y.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Rosen, E.D.; Walkey, C.J.; Puigserver, P.; Spiegelman, B.M. Transcriptional regulation of adipogenesis. Gene Dev. 2000, 14, 1293–1307. [Google Scholar] [CrossRef]
- Wang, W.J.; Bai, J.Y.; Liu, D.P.; Xue, L.M.; Zhu, X.Y. The antiinflammatory activity of shikonin and its inhibitory effect on leukotriene B4 biosynthesis. Yao Xue Xue Bao 1994, 29, 161–165. [Google Scholar]
- Lee, H.; Kang, R.; Yoon, Y. Shikonin inhibits fat accumulation in 3T3-L1 adipocytes. Phytother. Res. 2010, 24, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Han, H.; Lin, F.; Yang, L.; Feng, L.; Lai, X.; Wen, Z.; Yang, M.; Wang, C.; Ma, Y.; et al. Novel shikonin derivatives suppress cell proliferation, migration and induce apoptosis in human triple-negative breast cancer cells via regulating PDK1/PDHC axis. Life Sci. 2022, 310, 121077. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Jin, J.B.; Zhang, Z.Y.; Zuo, L.; Jiang, M.X.; Xie, C.F. Shikonin exerts antitumor activity by causing mitochondrial dysfunction in hepatocellular carcinoma through PKM2-AMPK-PGC1 alpha signaling pathway. Biochem. Cell. Biol. 2019, 97, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Gwon, S.Y.; Ahn, J.; Jung, C.H.; Moon, B.; Ha, T.Y. Shikonin Attenuates Hepatic Steatosis by Enhancing Beta Oxidation and Energy Expenditure via AMPK Activation. Nutrients 2020, 12, 1133. [Google Scholar] [CrossRef] [Green Version]
- Park, U.H.; Jeong, H.S.; Jo, E.Y.; Park, T.; Yoon, S.K.; Kim, E.J.; Jeong, J.C.; Um, S.J. Piperine, a component of black pepper, inhibits adipogenesis by antagonizing PPARgamma activity in 3T3-L1 cells. J. Agric. Food Chem. 2012, 60, 3853–3860. [Google Scholar] [CrossRef]
- Park, U.H.; Kang, M.R.; Kim, E.J.; Kwon, Y.S.; Hur, W.; Yoon, S.K.; Song, B.J.; Park, J.H.; Hwang, J.T.; Jeong, J.C.; et al. ASXL2 promotes proliferation of breast cancer cells by linking ER alpha to histone methylation. Oncogene 2016, 35, 3742–3752. [Google Scholar] [CrossRef]
- Eggleton, J.S.; Jialal, I. Thiazolidinediones. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Marcy, T.R.; Britton, M.L.; Blevins, S.M. Second-generation thiazolidinediones and hepatotoxicity. Ann. Pharmacother. 2004, 38, 1419–1423. [Google Scholar] [CrossRef]
- Lehrke, M.; Lazar, M.A. The many faces of PPAR gamma. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Katoch, S.; Sharma, V.; Patial, V. Peroxisome proliferator-activated receptor gamma as a therapeutic target for hepatocellular carcinoma: Experimental and clinical scenarios. World J. Gastroenterol. 2022, 28, 3535–3554. [Google Scholar] [CrossRef]
- Liu, C.L.; Yuan, Y.T.; Zhou, J.; Hu, R.X.; Ji, L.X.; Jiang, G.H. Piperine ameliorates insulin resistance via inhibiting metabolic inflammation in monosodium glutamate-treated obese mice. BMC Endocr. Disord. 2020, 20, 152. [Google Scholar] [CrossRef]
- Choi, S.; Choi, Y.; Choi, Y.; Kim, S.; Jang, J.; Park, T. Piperine reverses high fat diet-induced hepatic steatosis and insulin resistance in mice. Food Chem. 2013, 141, 3627–3635. [Google Scholar] [CrossRef] [PubMed]
- Szallasi, A. Capsaicin for Weight Control: “Exercise in a Pill” (or Just Another Fad)? Pharmaceuticals 2022, 15, 851. [Google Scholar] [CrossRef] [PubMed]
- Gwon, S.; Choi, W.; Lee, D.; Ahn, J.; Jung, C.; Moon, B.; Ha, T. Shikonin protects against obesity through the modulation of adipogenesis, lipogenesis, and β-oxidation in vivo. J. Funct. Foods 2015, 16, 484–493. [Google Scholar] [CrossRef]
- Boulos, J.C.; Rahama, M.; Hegazy, M.-E.F.; Efferth, T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019, 459, 248–267. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, U.-H.; Youn, H.; Kim, E.-J.; Um, S.-J. Shikonin Binds and Represses PPARγ Activity by Releasing Coactivators and Modulating Histone Methylation Codes. Nutrients 2023, 15, 1797. https://doi.org/10.3390/nu15071797
Park U-H, Youn H, Kim E-J, Um S-J. Shikonin Binds and Represses PPARγ Activity by Releasing Coactivators and Modulating Histone Methylation Codes. Nutrients. 2023; 15(7):1797. https://doi.org/10.3390/nu15071797
Chicago/Turabian StylePark, Ui-Hyun, HyeSook Youn, Eun-Joo Kim, and Soo-Jong Um. 2023. "Shikonin Binds and Represses PPARγ Activity by Releasing Coactivators and Modulating Histone Methylation Codes" Nutrients 15, no. 7: 1797. https://doi.org/10.3390/nu15071797