

Focusing on Phosphorus Loads: From Healthy People to Chronic Kidney Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Distribution of Phosphorus
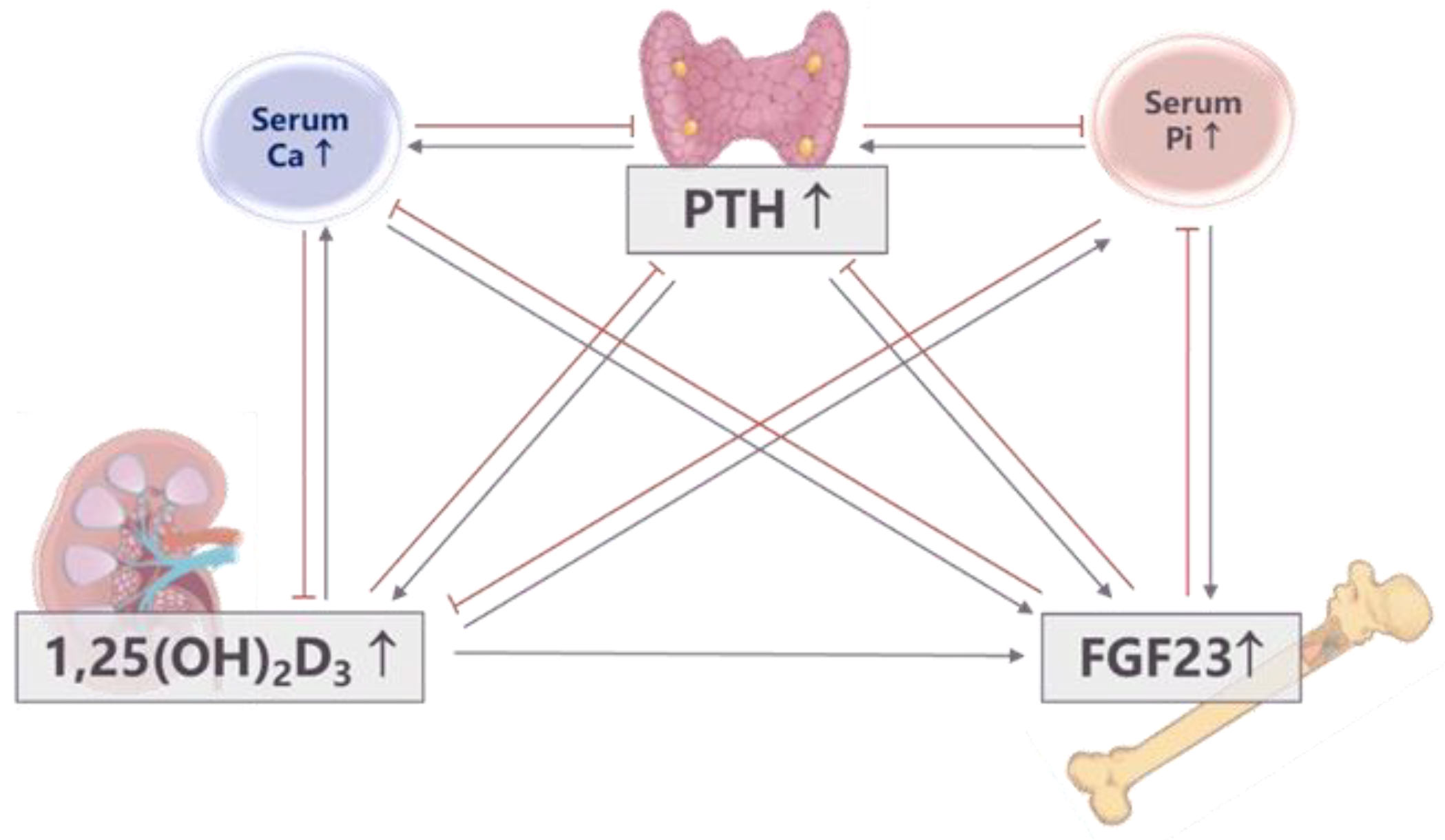
2. Phosphorus Homeostasis and In-Depth Review of Phosphorus Regulation
3. Phosphorus Pool
4. Phosphorus Overload and Pathological Phosphorus Pool
5. Reasons for and Deleterious Side Effects of Phosphorus Overload
6. Early Markers of Phosphorus Overload
7. The Path Forward
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, M.C.; Moe, O.W. Phosphate and Cellular Senescence. Adv. Exp. Med. Biol. 2022, 1362, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Naderi, A.S.A.; Reilly, R.F. Hereditary disorders of renal phosphate wasting. Nat. Rev. Nephrol. 2010, 6, 657–665. [Google Scholar] [CrossRef]
- Farrow, E.G.; White, K.E. Recent advances in renal phosphate handling. Nat. Rev. Nephrol. 2010, 6, 207–217. [Google Scholar] [CrossRef]
- Penido, M.G.; Alon, U.S. Phosphate homeostasis and its role in bone health. Pediatr. Nephrol. 2012, 27, 2039–2048. [Google Scholar] [CrossRef] [Green Version]
- Peacock, M. Phosphate Metabolism in Health and Disease. Calcif. Tissue Int. 2021, 108, 3–15. [Google Scholar] [CrossRef]
- Marks, J.; Debnam, E.S.; Unwin, R.J. Phosphate homeostasis and the renal-gastrointestinal axis. Am. J. Physiol. Renal. Physiol. 2010, 299, F285–F296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peacock, M. Hypoparathyroidism and the Kidney. Endocrinol. Metab. Clin. N. Am. 2018, 47, 839–853. [Google Scholar] [CrossRef]
- Martin, D.R.; Ritter, C.S.; Slatopolsky, E.; Brown, A.J. Acute regulation of parathyroid hormone by dietary phosphate. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E729–E734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, S.L.; Bonjour, J.P.; Rizzoli, R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J. Clin. Endocrinol. Metab. 2005, 90, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Bargagli, M.; Arena, M.; Naticchia, A.; Gambaro, G.; Mazzaferro, S.; Fuster, D.; Ferraro, P.M. The Role of Diet in Bone and Mineral Metabolism and Secondary Hyperparathyroidism. Nutrients 2021, 13, 2328. [Google Scholar] [CrossRef]
- Ito, N.; Fukumoto, S.; Takeuchi, Y.; Takeda, S.; Suzuki, H.; Yamashita, T.; Fujita, T. Effect of acute changes of serum phosphate on fibroblast growth factor (FGF)23 levels in humans. J. Bone Miner. Metab. 2007, 25, 419–422. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner Res. 2004, 19, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef]
- Yoda, K.; Imanishi, Y.; Yoda, M.; Mishima, T.; Ichii, M.; Yamada, S.; Mori, K.; Emoto, M.; Inaba, M. Impaired response of FGF-23 to oral phosphate in patients with type 2 diabetes: A possible mechanism of atherosclerosis. J. Clin. Endocrinol. Metab. 2012, 97, E2036–E2043. [Google Scholar] [CrossRef]
- Krajisnik, T.; Bjorklund, P.; Marsell, R.; Ljunggren, O.; Akerstrom, G.; Jonsson, K.B.; Westin, G.; Larsson, T.E. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J. Endocrinol. 2007, 195, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Kolek, O.I.; Hines, E.R.; Jones, M.D.; LeSueur, L.K.; Lipko, M.A.; Kiela, P.R.; Collins, J.F.; Haussler, M.R.; Ghishan, F.K. 1alpha,25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: The final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1036–G1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, Y.; Bivi, N.; Farrow, E.; Lezcano, V.; Plotkin, L.I.; White, K.E.; Bellido, T. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 2011, 49, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenza, H.L.; DeLuca, H.F. Regulation of 25-hydroxyvitamin D3 1alpha-hydroxylase gene expression by parathyroid hormone and 1,25-dihydroxyvitamin D3. Arch. Biochem. Biophys 2000, 381, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Shi, M.; Moe, O.W. Role of alphaKlotho and FGF23 in regulation of type II Na-dependent phosphate co-transporters. Pflugers Arch. 2019, 471, 99–108. [Google Scholar] [CrossRef]
- Sugihara, G.; May, R.; Ye, H.; Hsieh, C.H.; Deyle, E.; Fogarty, M.; Munch, S. Detecting causality in complex ecosystems. Science 2012, 338, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Miller, K.L.; Salimi-Khorshidi, G.; Webster, M.; Beckmann, C.F.; Nichols, T.E.; Ramsey, J.D.; Woolrich, M.W. Network modelling methods for FMRI. Neuroimage 2011, 54, 875–891. [Google Scholar] [CrossRef] [PubMed]
- Fornito, A.Z.A.; Bullmore, E.T. Fundamentals of Brain Network Analysis, 1st ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Ye, G.; Zhang, J.; Bi, Z.; Zhang, W.; Zhang, M.; Zhang, Q.; Wang, M.; Chen, J. Dominant factors of the phosphorus regulatory network differ under various dietary phosphate loads in healthy individuals. Ren. Fail. 2021, 43, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Massy, Z.A.; Drueke, T.B. Strategies for Phosphate Control in Patients With CKD. Kidney Int. Rep. 2019, 4, 1043–1056. [Google Scholar] [CrossRef] [Green Version]
- Stremke, E.R.; Wiese, G.N.; Moe, S.M.; Wastney, M.E.; Moorthi, R.N.; Gallant, K.M.H. Intestinal Phosphorus Absorption in Moderate CKD and Healthy Adults Determined Using a Radioisotopic Tracer. J. Am. Soc. Nephrol. 2021, 32, 2057–2069. [Google Scholar] [CrossRef]
- Musso, C.G.; Gregori, J.A.A.; Macías-Núñez, J.F. Renal Handling of Uric Acid, Magnesium, Phosphorus, Calcium, and Acid Base in the Elderly. In The Aging Kidney in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2008; pp. 155–171. [Google Scholar]
- Nishida, Y.; Taketani, Y.; Yamanaka-Okumura, H.; Imamura, F.; Taniguchi, A.; Sato, T.; Shuto, E.; Nashiki, K.; Arai, H.; Yamamoto, H.; et al. Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int. 2006, 70, 2141–2147. [Google Scholar] [CrossRef] [Green Version]
- Scanni, R.; von Rotz, M.; Jehle, S.; Hulter, H.N.; Krapf, R. The human response to acute enteral and parenteral phosphate loads. J. Am. Soc. Nephrol. 2014, 25, 2730–2739. [Google Scholar] [CrossRef] [Green Version]
- Kemp, G.J.; Blumsohn, A.; Morris, B.W. Circadian changes in plasma phosphate concentration, urinary phosphate excretion, and cellular phosphate shifts. Clin. Chem. 1992, 38, 400–402. [Google Scholar] [CrossRef]
- Markowitz, M.; Rotkin, L.; Rosen, J.F. Circadian rhythms of blood minerals in humans. Science 1981, 213, 672–674. [Google Scholar] [CrossRef]
- Portale, A.A.; Halloran, B.P.; Morris, R.C., Jr. Dietary intake of phosphorus modulates the circadian rhythm in serum concentration of phosphorus. Implications for the renal production of 1,25-dihydroxyvitamin D. J. Clin. Invest. 1987, 80, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Ix, J.H.; Anderson, C.A.; Smits, G.; Persky, M.S.; Block, G.A. Effect of dietary phosphate intake on the circadian rhythm of serum phosphate concentrations in chronic kidney disease: A crossover study. Am. J. Clin. Nutr. 2014, 100, 1392–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsmith, R.S.; Siemsen, A.W.; Mason, A.D., Jr.; Forland, M. Primary role of plasma hydrocortisone concentration in the regulation of the normal forenoon pattern of urinary phosphate excretion. J. Clin. Endocrinol. Metab. 1965, 25, 1649–1659. [Google Scholar] [CrossRef]
- Kitamura, N.; Shigeno, C.; Shiomi, K.; Lee, K.; Ohta, S.; Sone, T.; Katsushima, S.; Tadamura, E.; Kousaka, T.; Yamamoto, I.; et al. Episodic fluctuation in serum intact parathyroid hormone concentration in men. J. Clin. Endocrinol. Metab. 1990, 70, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Spalding, E.M.; Chamney, P.W.; Farrington, K. Phosphate kinetics during hemodialysis: Evidence for biphasic regulation. Kidney Int. 2002, 61, 655–667. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Li, H.; Liao, H.; Yu, Y.; You, L.; Zhu, J.; Huang, B.; Yuan, L.; Hao, C.; Chen, J. Phosphate removal model: An observational study of low-flux dialyzers in conventional hemodialysis therapy. Hemodial. Int. 2012, 16, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Agar, B.U.; Akonur, A.; Lo, Y.C.; Cheung, A.K.; Leypoldt, J.K. Kinetic model of phosphorus mobilization during and after short and conventional hemodialysis. Clin. J. Am. Soc. Nephrol. 2011, 6, 2854–2860. [Google Scholar] [CrossRef] [Green Version]
- Daugirdas, J.T. A two-pool kinetic model predicts phosphate concentrations during and shortly following a conventional (three times weekly) hemodialysis session. Nephrol. Dial. Transplant 2018, 33, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; You, H.Z.; Wang, M.J.; Zhang, Q.; Dong, X.Y.; Liu, J.F.; Chen, J. High-phosphorus diet controlled for sodium elevates blood pressure in healthy adults via volume expansion. J. Clin. Hypertens 2021, 23, 849–859. [Google Scholar] [CrossRef]
- Chazot, G.; Lemoine, S.; Kocevar, G.; Kalbacher, E.; Sappey-Marinier, D.; Rouviere, O.; Juillard, L. Intracellular Phosphate and ATP Depletion Measured by Magnetic Resonance Spectroscopy in Patients Receiving Maintenance Hemodialysis. J. Am. Soc. Nephrol. 2021, 32, 229–237. [Google Scholar] [CrossRef]
- Bevington, A.; Mundy, K.I.; Yates, A.J.; Kanis, J.A.; Russell, R.G.; Taylor, D.J.; Rajagopalan, B.; Radda, G.K. A study of intracellular orthophosphate concentration in human muscle and erythrocytes by 31P nuclear magnetic resonance spectroscopy and selective chemical assay. Clin. Sci. 1986, 71, 729–735. [Google Scholar] [CrossRef] [Green Version]
- Fiaccadori, E.; Coffrini, E.; Fracchia, C.; Rampulla, C.; Montagna, T.; Borghetti, A. Hypophosphatemia and phosphorus depletion in respiratory and peripheral muscles of patients with respiratory failure due to COPD. Chest 1994, 105, 1392–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minutolo, R.; Bellizzi, V.; Cioffi, M.; Iodice, C.; Giannattasio, P.; Andreucci, M.; Terracciano, V.; Di Iorio, B.R.; Conte, G.; De Nicola, L. Postdialytic rebound of serum phosphorus: Pathogenetic and clinical insights. J. Am. Soc. Nephrol. 2002, 13, 1046–1054. [Google Scholar] [CrossRef]
- Hruska, K.A.; Mathew, S.; Lund, R.J.; Memon, I.; Saab, G. The pathogenesis of vascular calcification in the chronic kidney disease mineral bone disorder: The links between bone and the vasculature. Semin. Nephrol. 2009, 29, 156–165. [Google Scholar] [CrossRef] [Green Version]
- Isakova, T.; Ix, J.H.; Sprague, S.M.; Raphael, K.L.; Fried, L.; Gassman, J.J.; Raj, D.; Cheung, A.K.; Kusek, J.W.; Flessner, M.F.; et al. Rationale and Approaches to Phosphate and Fibroblast Growth Factor 23 Reduction in CKD. J. Am. Soc. Nephrol. 2015, 26, 2328–2339. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Oka, M.; Maesato, K.; Ikee, R.; Mano, T.; Hidekazu, M.; Ohtake, T. Coronary artery calcification, ADMA, and insulin resistance in CKD patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 1289–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksen, B.O.; Palsson, R.; Ebert, N.; Melsom, T.; van der Giet, M.; Gudnason, V.; Indridason, O.S.; Inker, L.A.; Jenssen, T.G.; Levey, A.S.; et al. GFR in Healthy Aging: An Individual Participant Data Meta-Analysis of Iohexol Clearance in European Population-Based Cohorts. J. Am. Soc. Nephrol. 2020, 31, 1602–1615. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.A.; Viswanathan, G.; Weiner, D.E. Chronic kidney disease and end-stage renal disease in the elderly population: Current prevalence, future projections, and clinical significance. Adv. Chronic Kidney Dis. 2010, 17, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaoka, D.; Inaba, M.; Imanishi, Y.; Hayashi, N.; Ohara, M.; Nagata, Y.; Kurajoh, M.; Yamada, S.; Mori, K.; Emoto, M. Denosumab Improves Glomerular Filtration Rate in Osteoporotic Patients With Normal Kidney Function by Lowering Serum Phosphorus. J. Bone Miner. Res. 2019, 34, 2028–2035. [Google Scholar] [CrossRef]
- Sarafrazi, N.; Wambogo, E.A.; Shepherd, J.A. Osteoporosis or Low Bone Mass in Older Adults: United States, 2017–2018; CDC Stacks: Atlanta, GA, USA, 2021; No. 405. [Google Scholar]
- Mohammad, J.; Scanni, R.; Bestmann, L.; Hulter, H.N.; Krapf, R. A Controlled Increase in Dietary Phosphate Elevates BP in Healthy Human Subjects. J. Am. Soc. Nephrol. 2018, 29, 2089–2098. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.T.; Robinson-Cohen, C.; de Oliveira, M.C.; Kostina, A.; Nettleton, J.A.; Ix, J.H.; Nguyen, H.; Eng, J.; Lima, J.A.; Siscovick, D.S.; et al. Dietary phosphorus is associated with greater left ventricular mass. Kidney Int. 2013, 83, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.; Batch, B.C.; McGuire, H.L.; Vollmer, W.M.; Svetkey, L.P.; Tyson, C.C.; Sanguankeo, A.; Anderson, C.; Houston, J.; Appel, L.J. Association of a reduction in central obesity and phosphorus intake with changes in urinary albumin excretion: The PREMIER study. Am. J. Kidney Dis. 2013, 62, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Konig, M.; Stock, C.; Wiesinger, A.; Hillebrand, U.; Reiermann, S.; Reuter, S.; Amler, S.; Kohler, G.; Buck, F.; et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2013, 83, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Stevens, K.K.; Denby, L.; Patel, R.K.; Mark, P.B.; Kettlewell, S.; Smith, G.L.; Clancy, M.J.; Delles, C.; Jardine, A.G. Deleterious effects of phosphate on vascular and endothelial function via disruption to the nitric oxide pathway. Nephrol. Dial. Transplant 2017, 32, 1617–1627. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.R.; Anderson, C. Dietary Phosphorus Intake and the Kidney. Annu. Rev. Nutr. 2017, 37, 321–346. [Google Scholar] [CrossRef] [Green Version]
- Ibels, L.S.; Alfrey, A.C.; Haut, L.; Huffer, W.E. Preservation of function in experimental renal disease by dietary restriction of phosphate. N. Engl. J. Med. 1978, 298, 122–126. [Google Scholar] [CrossRef]
- Shiizaki, K.; Tsubouchi, A.; Miura, Y.; Seo, K.; Kuchimaru, T.; Hayashi, H.; Iwazu, Y.; Miura, M.; Battulga, B.; Ohno, N.; et al. Calcium phosphate microcrystals in the renal tubular fluid accelerate chronic kidney disease progression. J. Clin. Invest. 2021, 131, 16. [Google Scholar] [CrossRef]
- Ko, G.J.; Rhee, C.M.; Kalantar-Zadeh, K.; Joshi, S. The Effects of High-Protein Diets on Kidney Health and Longevity. J. Am. Soc. Nephrol. 2020, 31, 1667–1679. [Google Scholar] [CrossRef]
- Pinheiro, M.M.; Schuch, N.J.; Genaro, P.S.; Ciconelli, R.M.; Ferraz, M.B.; Martini, L.A. Nutrient intakes related to osteoporotic fractures in men and women--the Brazilian Osteoporosis Study (BRAZOS). Nutr. J. 2009, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.R.; Lazo, M.; Appel, L.J.; Gutierrez, O.M.; Grams, M.E. High dietary phosphorus intake is associated with all-cause mortality: Results from NHANES III. Am. J. Clin. Nutr. 2014, 99, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gomez, M.C.; Vilahur, G. Osteoporosis and vascular calcification: A shared scenario. Clin. Investig. Arterioscler. 2020, 32, 33–42. [Google Scholar] [CrossRef] [PubMed]
- von der Recke, P.; Hansen, M.A.; Hassager, C. The association between low bone mass at the menopause and cardiovascular mortality. Am. J. Med. 1999, 106, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Appelman-Dijkstra, N.M.; Papapoulos, S.E. Modulating Bone Resorption and Bone Formation in Opposite Directions in the Treatment of Postmenopausal Osteoporosis. Drugs 2015, 75, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruska, K.A.; Mathew, S.; Lund, R. Osteoporosis and cardiovascular disease: Lessons from chronic kidney disease. Clin. Cases Miner. Bone Metab. 2008, 5, 35–39. [Google Scholar]
- Cannata-Andia, J.B.; Carrillo-Lopez, N.; Messina, O.D.; Hamdy, N.A.T.; Panizo, S.; Ferrari, S.L.; On Behalf Of The International Osteoporosis Foundation Iof Working Group On Bone And Cardiovascular Diseases. Pathophysiology of Vascular Calcification and Bone Loss: Linked Disorders of Ageing? Nutrients 2021, 13, 835. [Google Scholar] [CrossRef]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef]
- Ginsberg, C.; Houben, A.; Malhotra, R.; Berendschot, T.; Dagnelie, P.C.; Kooman, J.P.; Webers, C.A.; Stehouwer, C.D.A.; Ix, J.H. Serum Phosphate and Microvascular Function in a Population-Based Cohort. Clin J. Am. Soc. Nephrol. 2019, 14, 1626–1633. [Google Scholar] [CrossRef]
- Foley, R.N.; Collins, A.J.; Herzog, C.A.; Ishani, A.; Kalra, P.A. Serum phosphorus levels associate with coronary atherosclerosis in young adults. J. Am. Soc. Nephrol. 2009, 20, 397–404. [Google Scholar] [CrossRef] [Green Version]
- O’Seaghdha, C.M.; Hwang, S.J.; Muntner, P.; Melamed, M.L.; Fox, C.S. Serum phosphorus predicts incident chronic kidney disease and end-stage renal disease. Nephrol. Dial. Transpl. 2011, 26, 2885–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, M.; Sacks, F.; Pfeffer, M.; Gao, Z.; Curhan, G.; Cholesterol And Recurrent Events (CARE) Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 2005, 112, 2627–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Li, H.; You, L.; Yu, X.; Zhang, M.; Zhu, R.; Hao, C.; Zhang, Z.; Chen, J. Association of serum phosphorus variability with coronary artery calcification among hemodialysis patients. PLoS ONE 2014, 9, e93360. [Google Scholar] [CrossRef] [Green Version]
- Komaba, H.; Fukagawa, M. Phosphate-a poison for humans? Kidney Int. 2016, 90, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, N.D.; Holt, S.G. Is serum phosphate a useful target in patients with chronic kidney disease and what is the role for dietary phosphate restriction? Nephrology 2017, 22 (Suppl. S2), 36–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bricker, N.S. On the pathogenesis of the uremic state. An exposition of the “trade-off hypothesis”. N. Engl. J. Med. 1972, 286, 1093–1099. [Google Scholar] [CrossRef]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutierrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [Green Version]
- Takashi, Y.; Kosako, H.; Sawatsubashi, S.; Kinoshita, Y.; Ito, N.; Tsoumpra, M.K.; Nangaku, M.; Abe, M.; Matsuhisa, M.; Kato, S.; et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc. Natl. Acad. Sci. USA 2019, 116, 11418–11427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, M.; Kinoshita, Y.; Taguchi, M.; Fukumoto, S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J. Bone Miner. Metab. 2016, 34, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Yamamoto, L.; Karaplis, A.C.; St-Arnaud, R.; Goltzman, D. Fibroblast Growth Factor 23 Regulation by Systemic and Local Osteoblast-Synthesized 1,25-Dihydroxyvitamin D. J. Am. Soc. Nephrol. 2017, 28, 586–597. [Google Scholar] [CrossRef] [Green Version]
- Meir, T.; Durlacher, K.; Pan, Z.; Amir, G.; Richards, W.G.; Silver, J.; Naveh-Many, T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014, 86, 1106–1115. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Ortiz, M.E.; Lopez, I.; Munoz-Castaneda, J.R.; Martinez-Moreno, J.M.; Ramirez, A.P.; Pineda, C.; Canalejo, A.; Jaeger, P.; Aguilera-Tejero, E.; Rodriguez, M.; et al. Calcium deficiency reduces circulating levels of FGF23. J. Am. Soc. Nephrol. 2012, 23, 1190–1197. [Google Scholar] [CrossRef] [Green Version]
- Edmonston, D.; Wolf, M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat. Rev. Nephrol. 2020, 16, 7–19. [Google Scholar] [CrossRef]
- Mace, M.L.; Gravesen, E.; Hofman-Bang, J.; Olgaard, K.; Lewin, E. Key role of the kidney in the regulation of fibroblast growth factor 23. Kidney Int. 2015, 88, 1304–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neyra, J.A.; Hu, M.C.; Moe, O.W. Klotho in Clinical Nephrology: Diagnostic and Therapeutic Implications. Clin. J. Am. Soc. Nephrol. 2020, 16, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Castaneda, J.R.; Herencia, C.; Pendon-Ruiz de Mier, M.V.; Rodriguez-Ortiz, M.E.; Diaz-Tocados, J.M.; Vergara, N.; Martinez-Moreno, J.M.; Salmeron, M.D.; Richards, W.G.; Felsenfeld, A.; et al. Differential regulation of renal Klotho and FGFR1 in normal and uremic rats. FASEB J. 2017, 31, 3858–3867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stremke, E.R.; McCabe, L.D.; McCabe, G.P.; Martin, B.R.; Moe, S.M.; Weaver, C.M.; Peacock, M.; Gallant, K.M.H. Twenty-Four-Hour Urine Phosphorus as a Biomarker of Dietary Phosphorus Intake and Absorption in CKD: A Secondary Analysis from a Controlled Diet Balance Study. Clin. J. Am. Soc. Nephrol. 2018, 13, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drueke, T.B. Bone biopsy in chronic kidney disease: Still an option? J. Bras. Nefrol. 2020, 42, 130–132. [Google Scholar] [CrossRef]
- Drueke, T.B.; Massy, Z.A. Changing bone patterns with progression of chronic kidney disease. Kidney Int. 2016, 89, 289–302. [Google Scholar] [CrossRef] [Green Version]
- Carbonare, L.D.; Valenti, M.T.; Giannini, S.; Gallieni, M.; Stefani, F.; Ciresa, R.; Politi, C.; Fusaro, M. Bone Biopsy for Histomorphometry in Chronic Kidney Disease (CKD): State-of-the-Art and New Perspectives. J. Clin. Med. 2021, 10, 4617. [Google Scholar] [CrossRef]
- Ye, X.; Kooman, J.P.; van der Sande, F.M.; Raimann, J.G.; Usvyat, L.A.; Wang, Y.; Maddux, F.W.; Kotanko, P. Relationship between serum phosphate levels and survival in chronic hemodialysis patients: Interactions with age, malnutrition and inflammation. Clin. Kidney J. 2021, 14, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Fouque, D.; Roth, H.; Pelletier, S.; London, G.M.; Hannedouche, T.; Jean, G.; Bouchet, J.L.; Drueke, T. Control of mineral metabolism and bone disease in haemodialysis patients: Which optimal targets? Nephrol. Dial. Transpl. 2013, 28, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.R.; Grams, M.E. Serum phosphorus and mortality in the Third National Health and Nutrition Examination Survey (NHANES III): Effect modification by fasting. Am. J. Kidney Dis. 2014, 64, 567–573. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Ye, G.; Bi, Z.; Chen, W.; Qian, J.; Zhang, M.; Ding, D.; Wang, M.; Chen, J. Higher one-year achievement rate of serum phosphate associated with lower cardiovascular mortality in hemodialysis patients. BMC Nephrol. 2021, 22, 398. [Google Scholar] [CrossRef] [PubMed]
- Barsotti, G.; Giannoni, A.; Morelli, E.; Lazzeri, M.; Vlamis, I.; Baldi, R.; Giovannetti, S. The decline of renal function slowed by very low phosphorus intake in chronic renal patients following a low nitrogen diet. Clin. Nephrol. 1984, 21, 54–59. [Google Scholar] [PubMed]
- Sigrist, M.; Tang, M.; Beaulieu, M.; Espino-Hernandez, G.; Er, L.; Djurdjev, O.; Levin, A. Responsiveness of FGF-23 and mineral metabolism to altered dietary phosphate intake in chronic kidney disease (CKD): Results of a randomized trial. Nephrol. Dial. Transpl. 2013, 28, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakova, T.; Barchi-Chung, A.; Enfield, G.; Smith, K.; Vargas, G.; Houston, J.; Xie, H.; Wahl, P.; Schiavenato, E.; Dosch, A.; et al. Effects of dietary phosphate restriction and phosphate binders on FGF23 levels in CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 1009–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, G.A.; Wheeler, D.C.; Persky, M.S.; Kestenbaum, B.; Ketteler, M.; Spiegel, D.M.; Allison, M.A.; Asplin, J.; Smits, G.; Hoofnagle, A.N.; et al. Effects of phosphate binders in moderate CKD. J. Am. Soc. Nephrol. 2012, 23, 1407–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chue, C.D.; Townend, J.N.; Moody, W.E.; Zehnder, D.; Wall, N.A.; Harper, L.; Edwards, N.C.; Steeds, R.P.; Ferro, C.J. Cardiovascular effects of sevelamer in stage 3 CKD. J. Am. Soc. Nephrol. 2013, 24, 842–852. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.E.; de las Fuentes, L.; Rothstein, M.; Dietzen, D.J.; Bierhals, A.J.; Cheng, S.C.; Ross, W.; Windus, D.; Davila-Roman, V.G.; Hruska, K.A. Effects of phosphate binder therapy on vascular stiffness in early-stage chronic kidney disease. Am. J. Nephrol. 2013, 38, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Bellasi, A. Pro: Should phosphate binders be used in chronic kidney disease stage 3-4? Nephrol. Dial. Transpl. 2016, 31, 184–188. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; You, L.; Li, H.; Lin, Y.; Zhang, Z.; Hao, C.; Chen, J. Association of circulating fibroblast growth factor-23 with renal phosphate excretion among hemodialysis patients with residual renal function. Clin. J. Am. Soc. Nephrol. 2013, 8, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, M.; Li, H.; Yu, P.; Yuan, L.; Hao, C.; Chen, J.; Kalantar-Zadeh, K. Association of initial twice-weekly hemodialysis treatment with preservation of residual kidney function in ESRD patients. Am. J. Nephrol. 2014, 40, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Wang, M.; Zhang, M.; Zhang, W.; Shi, J.; Weng, J.; Huang, B.; Kalantar-Zadeh, K.; Chen, J. Benefits of Incremental Hemodialysis Seen in a Historical Cohort Study. Ther. Clin. Risk Manag. 2021, 17, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Obi, Y.; Streja, E.; Rhee, C.M.; Ravel, V.; Amin, A.N.; Cupisti, A.; Chen, J.; Mathew, A.T.; Kovesdy, C.P.; Mehrotra, R.; et al. Incremental Hemodialysis, Residual Kidney Function, and Mortality Risk in Incident Dialysis Patients: A Cohort Study. Am. J. Kidney Dis. 2016, 68, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, A.T.; Fishbane, S.; Obi, Y.; Kalantar-Zadeh, K. Preservation of residual kidney function in hemodialysis patients: Reviving an old concept. Kidney Int. 2016, 90, 262–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Zhang, J.; Kalantar-Zadeh, K.; Chen, J. Focusing on Phosphorus Loads: From Healthy People to Chronic Kidney Disease. Nutrients 2023, 15, 1236. https://doi.org/10.3390/nu15051236
Wang M, Zhang J, Kalantar-Zadeh K, Chen J. Focusing on Phosphorus Loads: From Healthy People to Chronic Kidney Disease. Nutrients. 2023; 15(5):1236. https://doi.org/10.3390/nu15051236
Chicago/Turabian StyleWang, Mengjing, Jiaying Zhang, Kamyar Kalantar-Zadeh, and Jing Chen. 2023. "Focusing on Phosphorus Loads: From Healthy People to Chronic Kidney Disease" Nutrients 15, no. 5: 1236. https://doi.org/10.3390/nu15051236