
Exploring the Anti-Inflammatory Effect of Inulin by Integrating Transcriptomic and Proteomic Analyses in a Murine Macrophage Cell Model

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Inuline Solutions
2.2. Raw 267.4 Cell Culture
2.3. Anti-Inflammatory Assays in Raw 264.7
2.3.1. Quantification of Nitric Oxide (NO)
2.3.2. Quantification of Cytokines
2.3.3. Data Collection
2.4. Transcriptomic Analysis
2.4.1. RNA Extraction, Library Preparation, and Sequencing
2.4.2. RNA-Seq Assay and Analysis
2.4.3. Differential Expression Analysis
2.4.4. Functional and Pathway Enrichment Analysis
2.5. Proteomic Analysis
2.5.1. Total Protein Extraction
2.5.2. Sample Preparation for Liquid Chromatography–Mass Spectrometry (LC-MS) Analysis
2.5.3. diaPASEF LC-MS Analysis
2.5.4. Data Analysis
2.6. Validation of Specific Genes by Quantitative Real-Time PCR
3. Results
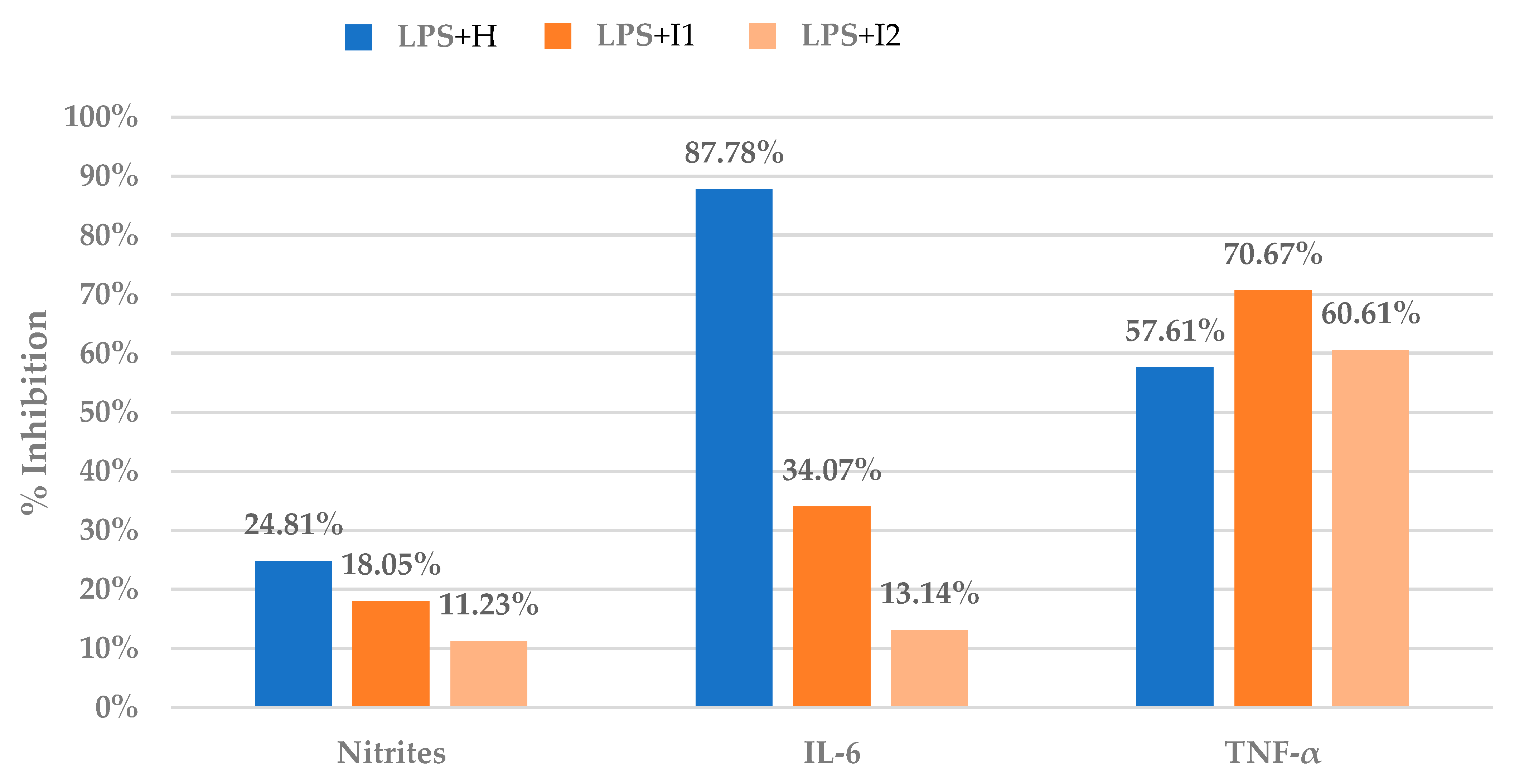
3.1. Anti-Inflammatory Assays in Raw 264.7
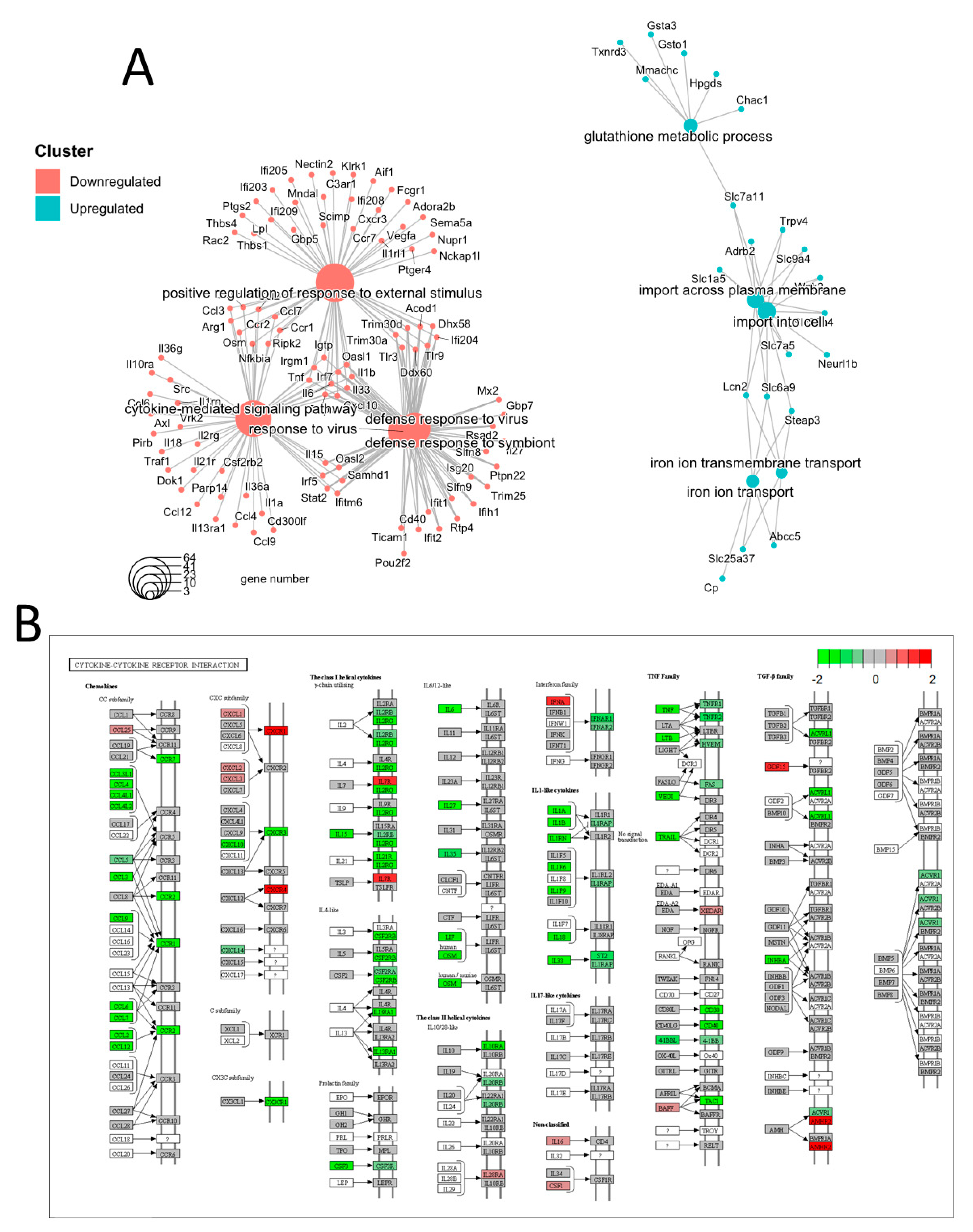
3.2. Transcriptomic Analysis
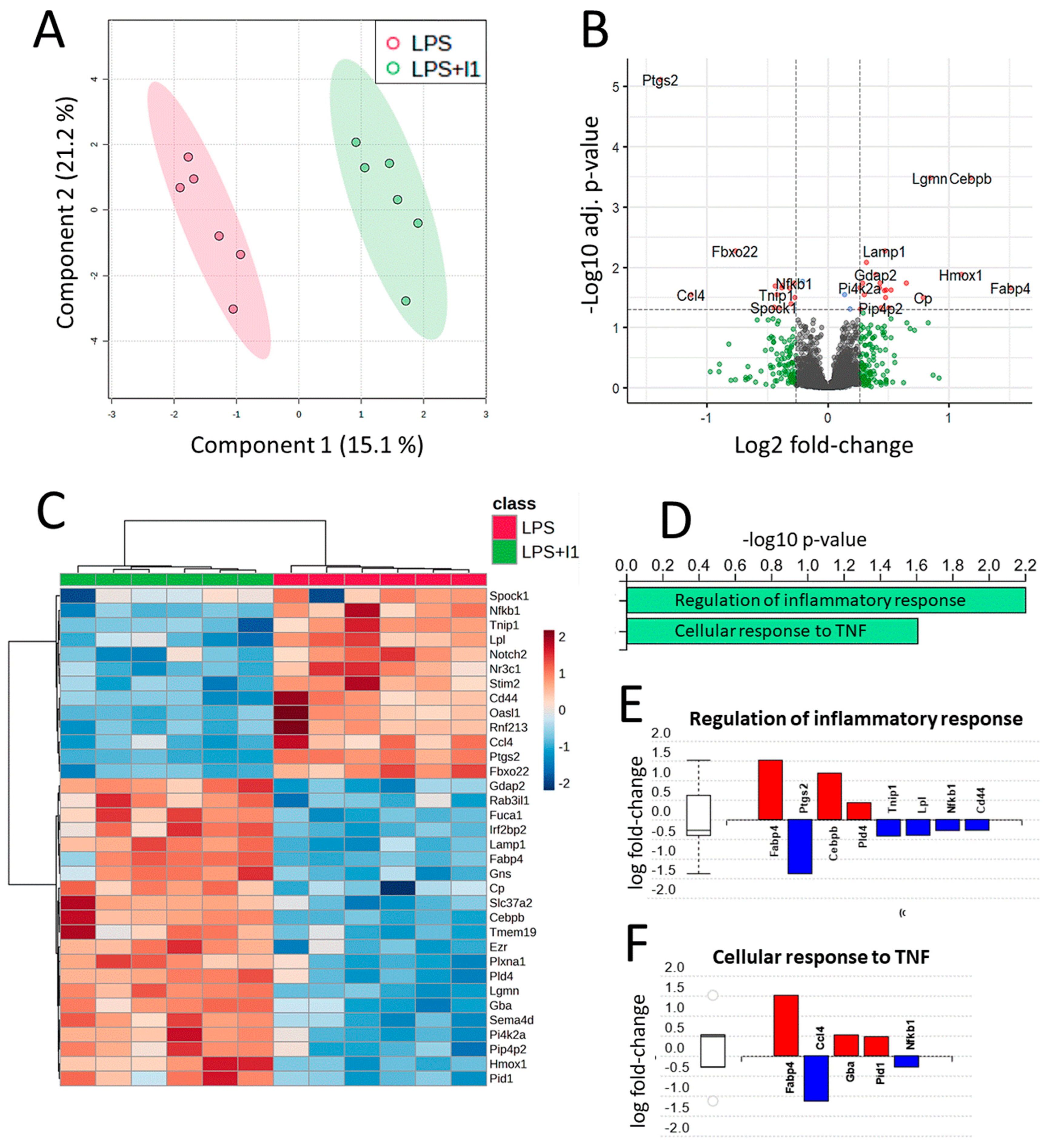
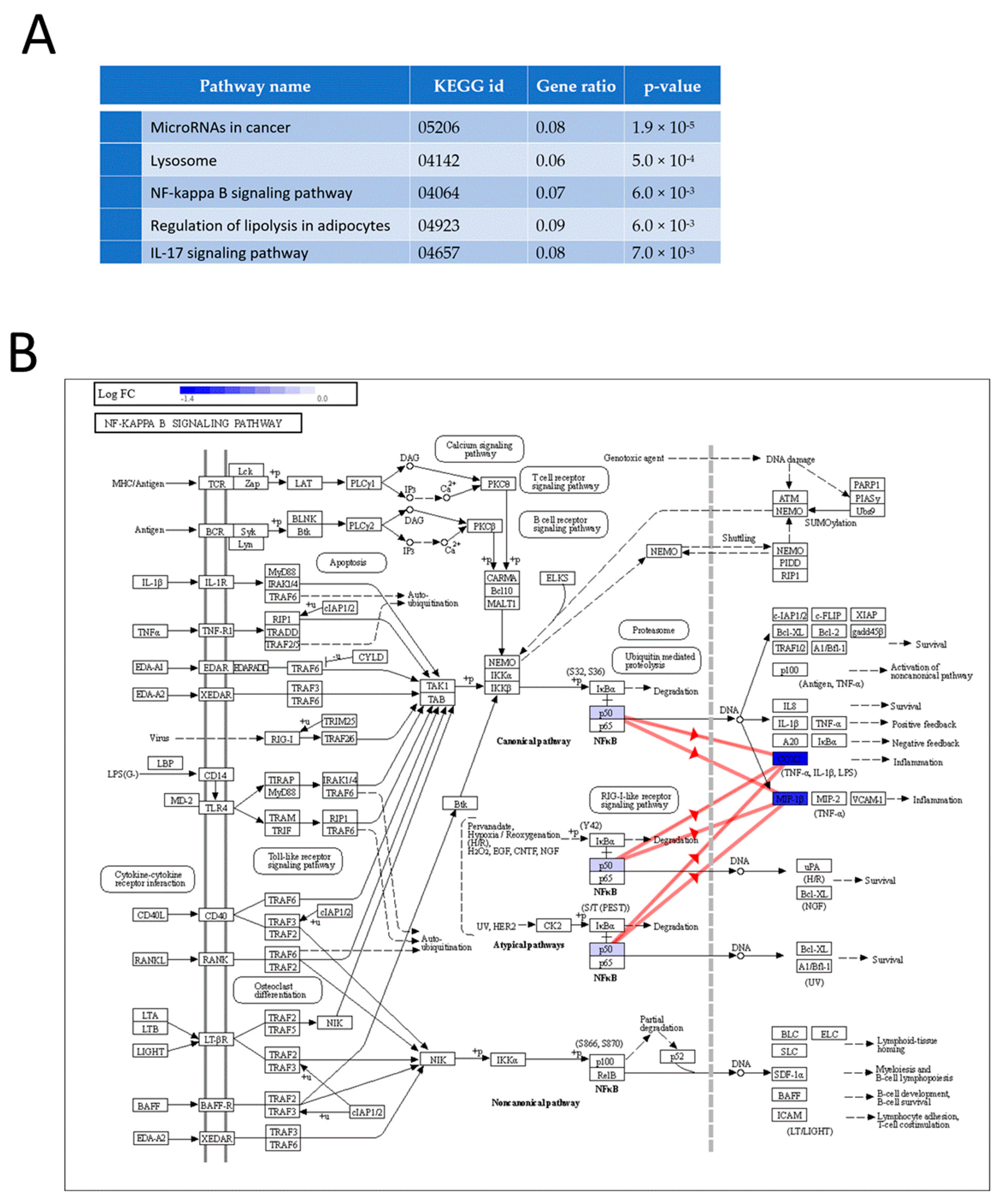
3.3. Proteomic Analysis
3.4. Validation of Selected Differentially Expressed Genes by qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gupta, N.; Jangid, A.K.; Pooja, D.; Kulhari, H. Inulin: A Novel and Stretchy Polysaccharide Tool for Biomedical and Nutritional Applications. Int. J. Biol. Macromol. 2019, 132, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Myhill, L.J.; Jensen, P.; Zakeri, A.; Nielsen, L.F.; Jakobsen, S.R.; Mejer, H.; Thamsborg, S.M.; Nejsum, P.; Williams, A.R. Effects of the Dietary Fibre Inulin and Trichuris Suis Products on Inflammatory Responses in Lipopolysaccharide-Stimulated Macrophages. Mol. Immunol. 2020, 121, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory Lymphocytes and Intestinal Inflammation. Annu. Rev. Immunol. 2009, 27, 313–338. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in Intestinal Inflammation and Resolution: A Potential Therapeutic Target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Hoentjen, F.; Welling, G.W.; Harmsen, H.J.M.; Zhang, X.; Snart, J.; Tannock, G.W.; Lien, K.; Churchill, T.A.; Lupicki, M.; Dieleman, L.A. Reduction of Colitis by Prebiotics in HLA-B27 Transgenic Rats Is Associated with Microflora Changes and Immunomodulation. Inflamm. Bowel Dis. 2005, 11, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Kehrl, J.H. Transforming Growth Factor-β: An Important Mediator of Immunoregulation. Int. J. Cell Cloning 1991, 9, 438–450. [Google Scholar] [CrossRef]
- Meng, Y.; Xu, Y.; Chang, C.; Qiu, Z.; Hu, J.; Wu, Y.; Zhang, B.; Zheng, G. Extraction, Characterization and Anti-Inflammatory Activities of an Inulin-Type Fructan from Codonopsis Pilosula. Int. J. Biol. Macromol. 2020, 163, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Moynagh, P.N. The NF-ΚB Pathway. J. Cell Sci. 2005, 118, 4589–4592. [Google Scholar] [CrossRef] [PubMed]
- Ortea, I. Foodomics in Health: Advanced Techniques for Studying the Bioactive Role of Foods. TrAC Trends Anal. Chem. 2022, 150, 116589. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Pandey, A.; Mann, M. Proteomics to Study Genes and Genomes. Nature 2000, 405, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Version 0.11.9. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 June 2021).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE Reference Annotation for the Human and Mouse Genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences [Version 2; Peer Review: 2 Approved]. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef]
- Morgan, M.; Shepherd, L. AnnotationHub: Client to Access AnnotationHub Resources. R Package Version 2.2.2. Available online: https://doi.org/doi:10.18129/B9.bioc.AnnotationHub (accessed on 1 June 2021).
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Ortea, I.; Gonzalez-Fernandez, M.J.; Ramos-Bueno, R.P.; Guil Guerrero, J.L. Proteomics Study Reveals That Docosahexaenoic and Arachidonic Acids Exert Different in vitro Anticancer Activities in Colorectal Cancer Cells. J. Agric. Food Chem. 2018, 66, 6003–6012. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.-D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. DiaPASEF: Parallel Accumulation–Serial Fragmentation Combined with Data-Independent Acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Spandidos, A.; Wang, X.; Wang, H.; Seed, B. PrimerBank: A Resource of Human and Mouse PCR Primer Pairs for Gene Expression Detection and Quantification. Nucleic Acids Res. 2010, 38, D792–D799. [Google Scholar] [CrossRef] [PubMed]
- Spandidos, A.; Wang, X.; Wang, H.; Dragnev, S.; Thurber, T.; Seed, B. A Comprehensive Collection of Experimentally Validated Primers for Polymerase Chain Reaction Quantitation of Murine Transcript Abundance. BMC Genom. 2008, 9, 633. [Google Scholar] [CrossRef]
- Wang, X.; Seed, B. A PCR Primer Bank for Quantitative Gene Expression Analysis. Nucleic Acids Res. 2003, 31, e154. [Google Scholar] [CrossRef]
- Hounkpe, B.W.; Chenou, F.; de Lima, F.; De Paula, E.V. HRT Atlas v1.0 Database: Redefining Human and Mouse Housekeeping Genes and Candidate Reference Transcripts by Mining Massive RNA-Seq Datasets. Nucleic Acids Res. 2021, 49, D947–D955. [Google Scholar] [CrossRef]
- Ruiz-Villalba, A.; Mattiotti, A.; Gunst, Q.D.; Cano-Ballesteros, S.; Van Den Hoff, M.J.; Ruijter, J.M. Reference Genes for Gene Expression Studies in the Mouse Heart. Sci. Rep. 2017, 7, 24. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. QBase Relative Quantification Framework and Software for Management and Automated Analysis of Real-Time Quantitative PCR Data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.-S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A Novel Signaling Pathway Impact Analysis. Bioinformatics 2009, 25, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and Chemokines: At the Crossroads of Cell Signalling and Inflammatory Disease. Biochim. Biophys. Acta—Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Malkowski, M.G. Substrate-Selective Inhibition of Cyclooxygeanse-2 by Fenamic Acid Derivatives Is Dependent on Peroxide Tone. J. Biol. Chem. 2016, 291, 15069–15081. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Marnett, L.J. Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Kakuta, S.; Shibata, S.; Iwakura, Y. Genomic Structure of the Mouse 2′,5′-Oligoadenylate Synthetase Gene Family. J. Interf. Cytokine Res. 2002, 22, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kim, B.; Oh, G.T.; Kim, Y.-J. OASL1 Inhibits Translation of the Type I Interferon–Regulating Transcription Factor IRF7. Nat. Immunol. 2013, 14, 346–355. [Google Scholar] [CrossRef]
- Chang, Y.; Kang, J.-S.; Jung, K.; Chung, D.H.; Ha, S.-J.; Kim, Y.-J.; Kim, H.Y. OASL1-Mediated Inhibition of Type I IFN Reduces Influenza A Infection-Induced Airway Inflammation by Regulating ILC2s. Allergy Asthma Immunol. Res. 2022, 14, 99–116. [Google Scholar] [CrossRef]
- Sim, C.K.; Cho, Y.S.; Kim, B.S.; Baek, I.-J.; Kim, Y.-J.; Lee, M.S. 2′–5′ Oligoadenylate Synthetase-like 1 (OASL1) Deficiency in Mice Promotes an Effective Anti-Tumor Immune Response by Enhancing the Production of Type I Interferons. Cancer Immunol. Immunother. 2016, 65, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Lee, M.S.; Kim, Y.-J.; Lee, H.K. OASL1 Deficiency Promotes Antiviral Protection against Genital Herpes Simplex Virus Type 2 Infection by Enhancing Type I Interferon Production. Sci. Rep. 2016, 6, 19089. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W. Heme Oxygenase-1: An Anti-Inflammatory Effector in Cardiovascular, Lung, and Related Metabolic Disorders. Antioxidants 2022, 11, 555. [Google Scholar] [CrossRef]
- Surh, Y.-J. Cancer Chemoprevention with Dietary Phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef]
- Mao, H.; Han, B.; Li, H.; Tao, Y.; Wu, W. FABP4 Knockdown Suppresses Inflammation, Apoptosis and Extracellular Matrix Degradation in IL-1β-Induced Chondrocytes by Activating PPARγ to Regulate the NF-ΚB Signaling Pathway. Mol. Med. Rep. 2021, 24, 855. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Shu, L.; Wu, X.; Liu, Y.; Cheong, L.Y.; Liao, B.; Xiao, X.; Hoo, R.L.C.; Zhou, Z.; Xu, A. Fatty Acid Binding Protein 4 Promotes Autoimmune Diabetes by Recruitment and Activation of Pancreatic Islet Macrophages. JCI Insight 2021, 6, e141814. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L.; Boord, J.B.; Maeda, K.; Babaev, V.R.; Uysal, K.T.; Morgan, M.A.; Parker, R.A.; Suttles, J.; Fazio, S.; Hotamisligil, G.S.; et al. Lack of Macrophage Fatty-Acid–Binding Protein AP2 Protects Mice Deficient in Apolipoprotein E against Atherosclerosis. Nat. Med. 2001, 7, 699–705. [Google Scholar] [CrossRef]
- Boord, J.B.; Maeda, K.; Makowski, L.; Babaev, V.R.; Fazio, S.; Linton, M.F.; Hotamisligil, G.S. Combined Adipocyte-Macrophage Fatty Acid–Binding Protein Deficiency Improves Metabolism, Atherosclerosis, and Survival in Apolipoprotein E–Deficient Mice. Circulation 2004, 110, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Fucho, R.; Görgün, C.Z.; Tuncman, G.; Cao, H.; Hotamisligil, G.S. Adipocyte/Macrophage Fatty Acid–Binding Proteins Contribute to Metabolic Deterioration through Actions in Both Macrophages and Adipocytes in Mice. J. Clin. Investig. 2008, 118, 2640–2650. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Y.; Zhan, S.; Zhang, W.; Xiong, F.; Ge, W. Comprehensive Proteome Analysis of Lysosomes Reveals the Diverse Function of Macrophages in Immune Responses. Oncotarget 2017, 8, 7420–7440. [Google Scholar] [CrossRef] [PubMed]
- Stow, J.L.; Ching Low, P.; Offenhäuser, C.; Sangermani, D. Cytokine Secretion in Macrophages and Other Cells: Pathways and Mediators. Immunobiology 2009, 214, 601–612. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Fold Change | p-Value | BH Adj. p-Value |
|---|---|---|---|
| Ptgs2 | 0.38 | 1.26 × 10−9 | 7.63 × 10−6 |
| Lgmn | 1.80 | 1.61 × 10−7 | 0.000325 |
| Cebpb | 2.27 | 1.54 × 10−7 | 0.000325 |
| Lamp1 | 1.39 | 4.45 × 10−6 | 0.005396 |
| Fbxo22 | 0.59 | 4.09 × 10−6 | 0.005396 |
| Fuca1 | 1.25 | 8.25 × 10−6 | 0.008340 |
| Gdap2 | 1.31 | 1.56 × 10−5 | 0.013015 |
| Hmox1 | 2.14 | 1.72 × 10−5 | 0.013015 |
| Nfkb1 | 0.82 | 3.76 × 10−5 | 0.018145 |
| Sema4d | 1.57 | 3.39 × 10−5 | 0.018145 |
| Irf2bp2 | 1.35 | 3.18 × 10−5 | 0.018145 |
| Ezr | 1.22 | 3.89 × 10−5 | 0.018145 |
| Nr3c1 | 0.77 | 4.62 × 10−5 | 0.020000 |
| Oasl1 | 0.74 | 5.10 × 10−5 | 0.020618 |
| Rnf213 | 0.77 | 6.01 × 10−5 | 0.021778 |
| Pi4k2a | 1.20 | 6.10 × 10−5 | 0.021778 |
| Pld4 | 1.34 | 6.82 × 10−5 | 0.021780 |
| Fabp4 | 2.85 | 6.81 × 10−5 | 0.021780 |
| Stim2 | 0.80 | 7.42 × 10−5 | 0.022509 |
| Gba | 1.43 | 8.42 × 10−5 | 0.023808 |
| Gns | 1.40 | 8.64 × 10−5 | 0.023808 |
| Plxna1 | 1.38 | 9.19 × 10−5 | 0.024230 |
| Slc37a2 | 1.23 | 1.27 × 10−4 | 0.028491 |
| Tnip1 | 0.75 | 1.26 × 10−4 | 0.028491 |
| Ccl4 | 0.46 | 1.19 × 10−4 | 0.028491 |
| Cp | 1.73 | 1.5 × 10−4 | 0.031392 |
| Cd44 | 0.83 | 1.49 × 10−4 | 0.031392 |
| Pid1 | 1.39 | 1.56 × 10−4 | 0.031473 |
| Notch2 | 0.81 | 2.06 × 10−4 | 0.040399 |
| Spock1 | 0.74 | 2.44 × 10−4 | 0.046330 |
| Pip4p2 | 1.36 | 2.64 × 10−4 | 0.047034 |
| Tmem192 | 1.42 | 2.71 × 10−4 | 0.047034 |
| Lpl | 0.76 | 2.63 × 10−4 | 0.047034 |
| Rab3il1 | 1.21 | 2.89 × 10−4 | 0.048628 |
| Protein/Gene Id | Name | Proteomics | Transcriptomics | qPCR | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| log2 FC | Adj. p-Value | Significant Change | log2 FC | Adj. p-Value | Significant Change | log2 FC | Adj. p-Value | Significant Change | ||
| Ptgs2 | Prostaglandin-endoperoxide synthase 2 | −1.38 | 7.6 × 10−6- | ↓ | −4.44 | 1.2 × 10−102 | ↓ | −0.05 | 1.4 × 10−3 | ↓ |
| Oasl1 | 2′-5′ oligoadenylate synthetase-like 1 | −0.43 | 2.1 × 10−2 | ↓ | −2.64 | 3.1 × 10−15 | ↓ | −0.16 | 2.2 × 10−3 | ↓ |
| Fabp4 | Fatty acid-binding protein 4 | 1.51 | 2.2 × 10−2 | ↑ | 4.05 | 1.7 × 10−91 | ↑ | 3.76 | 1.4 × 10−3 | ↑ |
| Slc37a2 | Solute carrier family 37 | 0.30 | 2.8 × 10−2 | ↑ | −1.16 | 4.2 × 10−4 | ↓ | −0.84 | 1.5 × 10−2 | ↓ |
| Cp | Ceruloplasmin | 0.79 | 3.1 × 10−2 | ↑ | 0.13 | 5.4 × 10−8 | ↑ | 0.59 | 1.0 × 10−2 | ↑ |
| Lpl | Lipoprotein lipase | −0.40 | 4.7 × 10−2 | ↓ | −2.19 | 3.4 × 10−26 | ↓ | −0.44 | 1.4 × 10−3 | ↓ |
| Il1a | Interleukin 1 alpha | n.d. | n.d. | −4.03 | 1.2 × 10−102 | ↓ | −0.11 | 1.3 × 10−3 | ↓ | |
| Il1b | Interleukin 1 beta | n.d. | n.d. | −6.28 | 9.4 × 10−31 | ↓ | −0.01 | 6.0 × 10−4 | ↓ | |
| Rgs16 | Regulator of G-protein signaling 16 | n.d. | n.d. | −5.37 | 4.4 × 10−92 | ↓ | −0.04 | 1.4 × 10−3 | ↓ | |
| Il6 | Interleukin 6 | n.d. | n.d. | −3.71 | 4.9 × 10−6 | ↓ | −0.15 | 4.0 × 10−3 | ↓ | |
| Il27 | Interleukin 27 | n.d. | n.d. | −4.02 | 8.1 × 10−72 | ↓ | −0.19 | 1.1 × 10−3 | ↓ | |
| Hvcn1 | Hydrogen voltage-gated channel 1 | n.d. | n.d. | 4.86 | 6.6 × 10−72 | ↑ | n.d. | n.d. | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farabegoli, F.; Santaclara, F.J.; Costas, D.; Alonso, M.; Abril, A.G.; Espiñeira, M.; Ortea, I.; Costas, C. Exploring the Anti-Inflammatory Effect of Inulin by Integrating Transcriptomic and Proteomic Analyses in a Murine Macrophage Cell Model. Nutrients 2023, 15, 859. https://doi.org/10.3390/nu15040859
Farabegoli F, Santaclara FJ, Costas D, Alonso M, Abril AG, Espiñeira M, Ortea I, Costas C. Exploring the Anti-Inflammatory Effect of Inulin by Integrating Transcriptomic and Proteomic Analyses in a Murine Macrophage Cell Model. Nutrients. 2023; 15(4):859. https://doi.org/10.3390/nu15040859
Chicago/Turabian StyleFarabegoli, Federica, Francisco J. Santaclara, Daniel Costas, Mercedes Alonso, Ana G. Abril, Montserrat Espiñeira, Ignacio Ortea, and Celina Costas. 2023. "Exploring the Anti-Inflammatory Effect of Inulin by Integrating Transcriptomic and Proteomic Analyses in a Murine Macrophage Cell Model" Nutrients 15, no. 4: 859. https://doi.org/10.3390/nu15040859