
Novel α-Glucosidase Inhibitory Peptides Identified In Silico from Dry-Cured Pork Loins with Probiotics through Peptidomic and Molecular Docking Analysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Dry-Cured Meat Products
2.2. Meat Protein Extraction and Hydrolysis
2.3. Peptidomic Characteristic
2.3.1. Peptide Identification by LC-MS/MS
2.3.2. α-Glucosidase Inhibitory Activity Peptides Search
2.3.3. Allergenic and ADMET Prediction
2.4. Molecular Docking
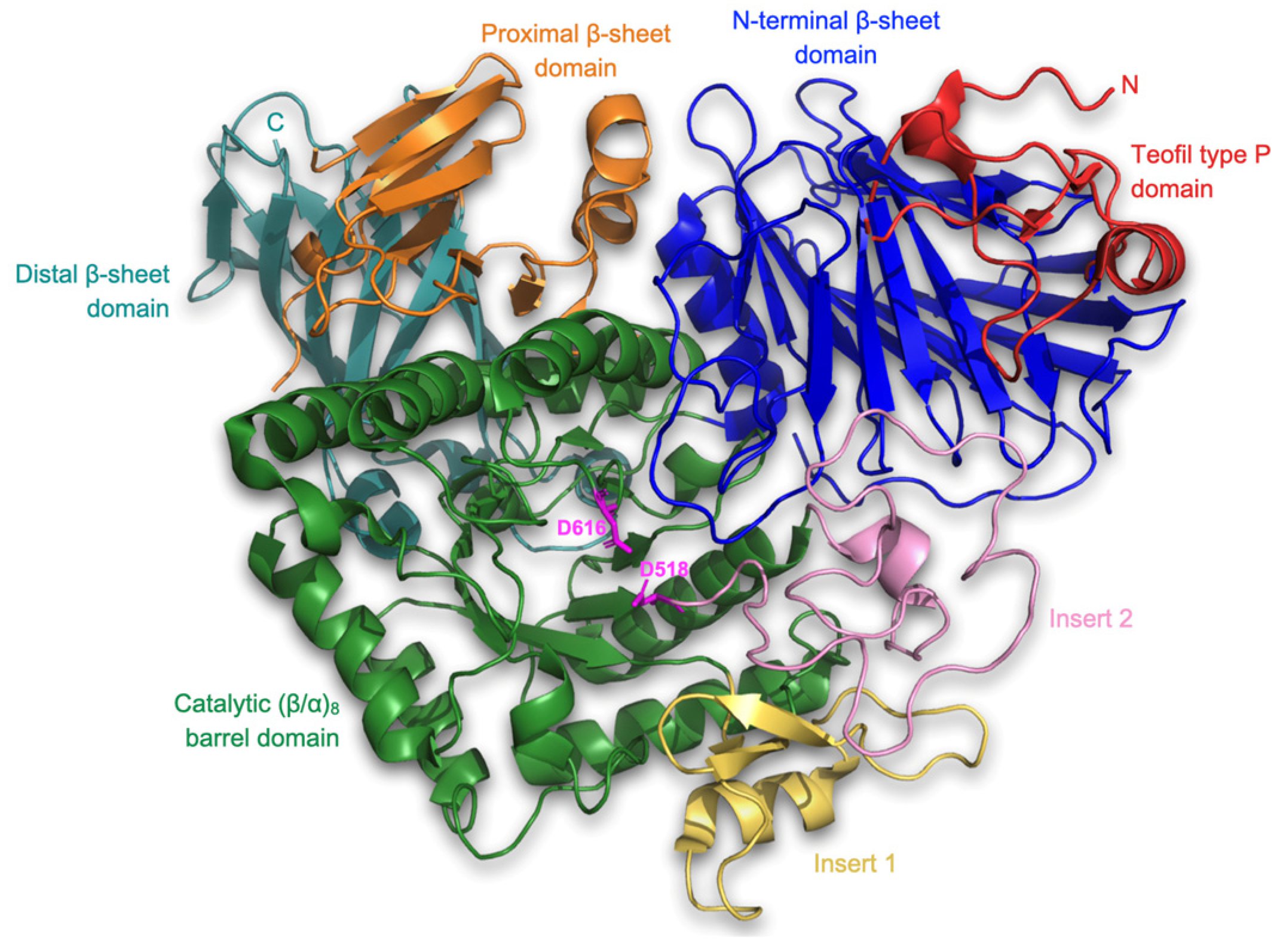
2.4.1. Receptor Structure and Preparation
2.4.2. Ligand Structures and Preparation
2.4.3. Molecular Docking Analysis
3. Results and Discussion
3.1. Peptide Characteristics
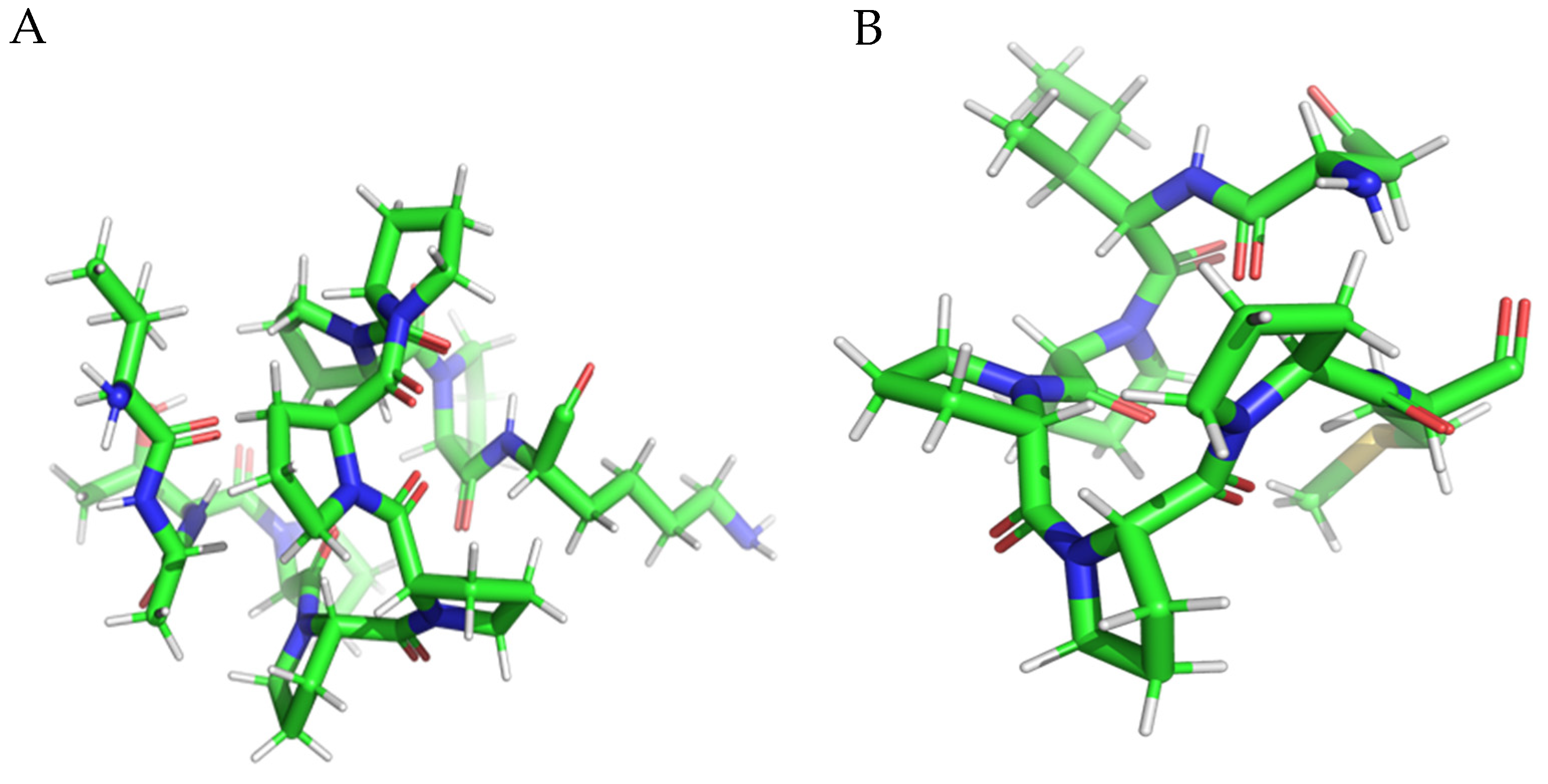
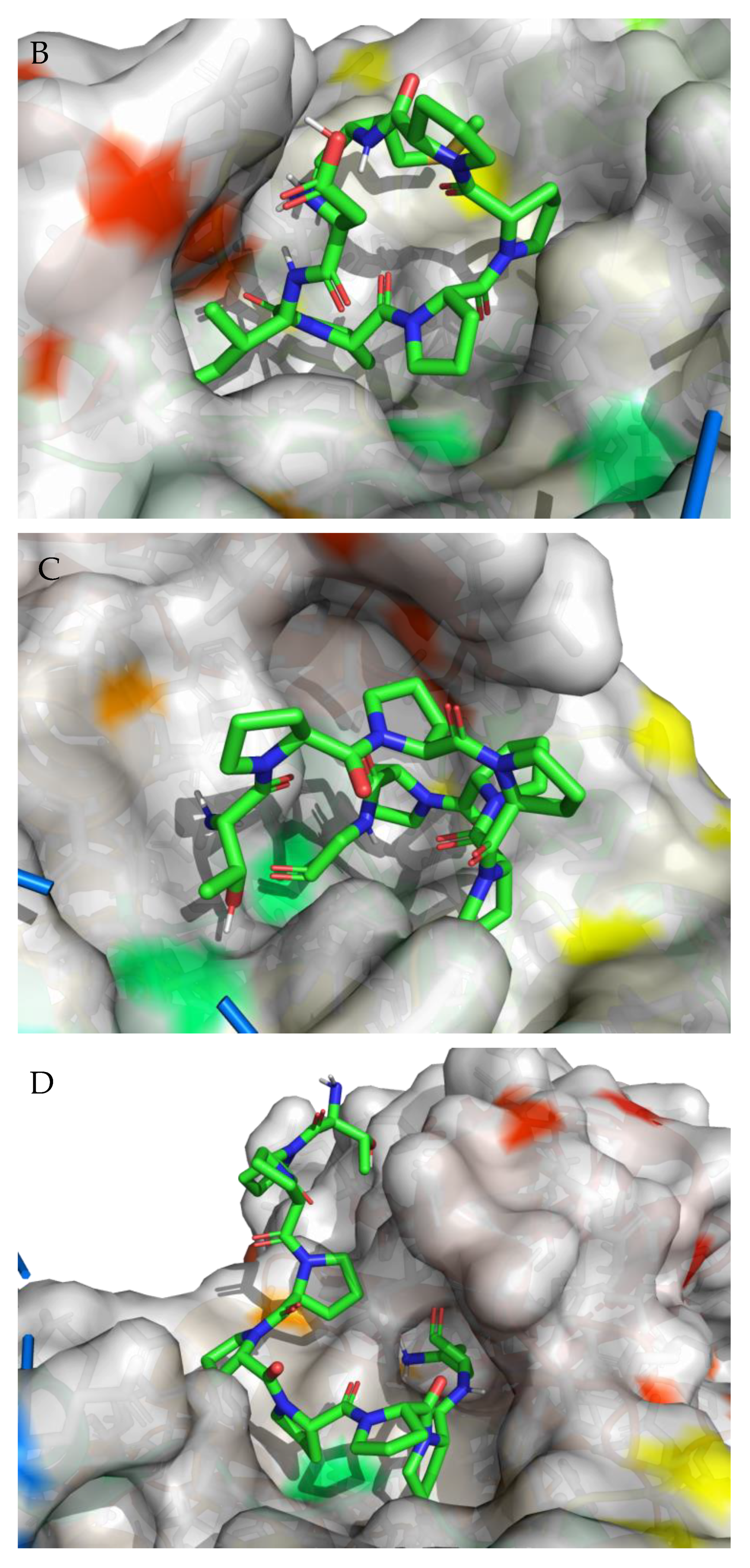
3.2. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Di Stefano, E.; Oliviero, T.; Udenigwe, C.C. Functional significance and structure–activity relationship of food-derived α-glucosidase inhibitors. Curr. Opin. Food Sci. 2018, 20, 7–12. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Bester, M.J.; Neitz, A.W.; Gaspar, A.R. Structural properties of bioactive peptides with α-glucosidase inhibitory activity. Chem. Biol. Drug Des. 2018, 91, 370–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Kongstad, K.T.; Wiese, S.; Jäger, A.K.; Staerk, D. Edible seaweed as future functional food: Identification of α-glucosidase inhibitors by combined use of high-resolution α-glucosidase inhibition profiling and HPLC–HRMS–SPE–NMR. Food Chem. 2016, 203, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Cisneros-Yupanqui, M.; Lante, A.; Mihaylova, D.; Krastanov, A.I.; Rizzi, C. The α-Amylase and α-Glucosidase Inhibition Capacity of Grape Pomace: A Review. Food Bioproc. Tech. 2022, 30, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Narwal, S.; Kumar, V.; Prakash, O. α-glucosidase inhibitors from plants: A natural approach to treat diabetes. Pharmacogn. Rev. 2011, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirir, A.M.; Daou, M.; Yousef, A.F.; Yousef, L.F. A review of alpha-glucosidase inhibitors from plants as potential candidates for the treatment of type-2 diabetes. Phytochem. Rev. 2022, 21, 1049–1079. [Google Scholar] [CrossRef]
- Martínez-Sánchez, S.M.; Minguela, A.; Prieto-Merino, D.; Zafrilla-Rentero, M.P.; Abellán-Alemán, J.; Montoro-García, S. The effect of regular intake of dry-cured ham rich in bioactive peptides on inflammation, platelet and monocyte activation markers in humans. Nutrients 2017, 9, 321. [Google Scholar] [CrossRef] [Green Version]
- Montoro-García, S.; Zafrilla-Rentero, M.P.; Celdrán-de Haro, F.M.; Piñero-de Armas, J.J.; Toldrá, F.; Tejada-Portero, L.; Abellan-Aleman, J. Effects of dry-cured ham rich in bioactive peptides on cardiovascular health: A randomized controlled trial. J. Funct. Foods 2017, 38, 160–167. [Google Scholar] [CrossRef]
- Kęska, P.; Stadnik, J. Potential DPP IV inhibitory peptides from dry-cured pork loins after hydrolysis: An in vitro and in silico study. Curr. Issues Mol. Biol. 2021, 43, 1335–1349. [Google Scholar] [CrossRef]
- Kęska, P.; Stadnik, J. Dipeptidyl Peptidase IV Inhibitory Peptides Generated in Dry-Cured Pork Loin during Aging and Gastrointestinal Digestion. Nutrients 2022, 14, 770. [Google Scholar] [CrossRef]
- Kęska, P.; Stadnik, J.; Bąk, O.; Borowski, P. Meat proteins as dipeptidyl peptidase iv inhibitors and glucose uptake stimulating peptides for the management of a type 2 diabetes mellitus in silico study. Nutrients 2019, 11, 2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kęska, P.; Stadnik, J. Structure–activity relationships study on biological activity of peptides as dipeptidyl peptidase IV inhibitors by chemometric modeling. Chem. Biol. Drug Des. 2020, 95, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Mora, L.; González-Rogel, D.; Heres, A.; Toldrá, F. Iberian dry-cured ham as a potential source of α-glucosidase-inhibitory peptides. J. Funct. Foods 2020, 67, 103840. [Google Scholar] [CrossRef]
- Molina, I.; Toldrá, F. Detection of proteolytic activity in microorganisms isolated from dry-cured ham. J. Food Sci. 1992, 57, 1308–1310. [Google Scholar] [CrossRef]
- Fadda, S.; Sanz, Y.; Vignolo, G.; Aristoy, M.C.; Oliver, G.; Toldrá, F. Characterization of muscle sarcoplasmic and myofibrillar protein hydrolysis caused by Lactobacillus plantarum. Appl. Environ. Microbiol. 1999, 65, 3540–3546. [Google Scholar] [CrossRef] [PubMed]
- Escudero, E.; Mora, L.; Toldrá, F. Stability of ACE inhibitory ham peptides against heat treatment and in vitro digestion. Food Chem. 2014, 161, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt KB. Available online: www.uniprot.org (accessed on 10 April 2023).
- BIOPEP-UWM. Available online: https://biochemia.uwm.edu.pl/biopep-uwm (accessed on 10 April 2023).
- ADMET. Available online: https://admet.scbdd.com (accessed on 10 April 2023).
- AllerTOP. Available online: https://www.ddg-pharmfac.net/AllerTOP/ (accessed on 10 April 2023).
- Hu, J.; Lai, X.; Wu, X.; Wang, H.; Weng, N.; Lu, J.; Lyu, M.; Wang, S. Isolation of a Novel Anti-Diabetic α-Glucosidase Oligo-Peptide Inhibitor from Fermented Rice Bran. Foods 2023, 12, 183. [Google Scholar] [CrossRef]
- Wairata, J.; Sukandar, E.R.; Fadlan, A.; Purnomo, A.S.; Taher, M.; Ersam, T. Evaluation of the antioxidant, antidiabetic, and antiplasmodial activities of xanthones isolated from Garcinia forbesii and their in silico studies. Biomedicines 2021, 9, 1380. [Google Scholar] [CrossRef]
- Roig-Zamboni, V.; Cobucci-Ponzano, B.; Iacono, R.; Ferrara, M.C.; Germany, S.; Bourne, Y.; Parenti, G.; Moracci, M.; Sulzenbacher, G. Structure of human lysosomal acid α-glucosidase—A guide for the treatment of Pompe disease. Nat. Commun. 2017, 8, 1111. [Google Scholar] [CrossRef] [Green Version]
- Sanner, M. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Model. 1999, 17, 57. [Google Scholar]
- Morris, G.; Huey, R.; Lindstrom, W.; Sanner, M.; Belew, R.; Goodsell, D.; Olson, A. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G. Open Babel: An open chemical toolbox. J. Chem. Inform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, N.M.; Alhossary, A.A.; Mu, Y.; Kwoh, C.-K. Protein-Ligand Blind Docking Using QuickVina-W With Inter-Process Spatio-Temporal Integration. Sci. Rep. 2017, 7, 15451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnautova, Y.A.; Jagielska, A.; Scheraga, H.A. A new force field (ECEPP-05) for peptides, proteins, and organic molecules. J. Phys. Chem. B 2006, 110, 5025–5044. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Yuan, Y.; Pei, J.; Lai, L. CAVITY: Mapping the Druggable Binding Site. In Computer-Aided Drug Discovery. Methods in Pharmacology and Toxicology; Zhang, W., Ed.; Humana Press: New York, NY, USA, 2015. [Google Scholar]
- Kawabata, T. Detection of multiscale pockets on protein surfaces using mathematical morphology. Proteins 2010, 78, 1195. [Google Scholar] [CrossRef]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM database of bioactive peptides: Current opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef] [Green Version]
- Kęska, P.; Stadnik, J. Peptidomic Characteristic of Peptides Generated in Dry-Cured Loins with Probiotic Strains of LAB during 360-Days Aging. Appl. Sci. 2022, 12, 6036. [Google Scholar] [CrossRef]
- Arámburo-Gálvez, J.G.; Arvizu-Flores, A.A.; Cárdenas-Torres, F.I.; Cabrera-Chávez, F.; Ramírez-Torres, G.I.; Flores-Mendoza, L.K.; Gastelum-Acosta, P.E.; Figueroa-Salcido, O.G.; Ontiveros, N. Prediction of ACE-I inhibitory peptides derived from chickpea (Cicer arietinum L.): In silico assessments using simulated enzymatic hydrolysis, molecular docking and ADMET evaluation. Foods 2022, 11, 1576. [Google Scholar] [CrossRef]
- Barrero, J.A.; Cabrera, F.; Cruz, C.M. Gliptins vs. Milk-derived Dipeptidyl-Peptidase IV Inhibiting Biopeptides: Physicochemical Characterization and Pharmacokinetic Profiling. Vitae 2021, 28, 346531. [Google Scholar] [CrossRef]
- Borawska-Dziadkiewicz, J.; Darewicz, M.; Tarczyńska, A.S. Properties of peptides released from salmon and carp via simulated human-like gastrointestinal digestion described applying quantitative parameters. PLoS ONE 2021, 16, e0255969. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Goel, N.; Yadav, T.C.; Pruthi, V. Quantum chemical, ADMET and molecular docking studies of ferulic acid amide derivatives with a novel anticancer drug target. Med. Chem. Res. 2017, 26, 1822–1834. [Google Scholar] [CrossRef]
- Nisha, C.M.; Kumar, A.; Nair, P.; Gupta, N.; Silakari, C.; Tripathi, T.; Kumar, A. Molecular docking and in silico ADMET study reveals acylguanidine 7a as a potential inhibitor of β-secretase. Adv. Bioinform. 2016, 2016, 9258578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altuner, E.M. In Silico Proof of the Effect of Quercetin and Umbelliferone as Alpha-Amylase Inhibitors, Which Can Be Used in the Treatment of Diabetes. Kastamonu Univ. J. For. 2022, 22, 202–216. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Mamadalieva, N.Z.; Youssef, F.S.; Hussain, H.; Zengin, G.; Mollica, A.; Al Musayeib, N.M.; Ashour, M.L.; Westermann, B.; Wessjohann, L.A. Validation of the antioxidant and enzyme inhibitory potential of selected triterpenes using in vitro and in silico studies, and the evaluation of their ADMET properties. Molecules 2021, 26, 6331. [Google Scholar] [CrossRef]
- Duchin, K.L.; McKinstry, D.N.; Cohen, A.I.; Migdalof, B.H. Pharmacokinetics of captopril in healthy subjects and in patients with cardiovascular diseases. Clin. Pharmacokinet. 1988, 14, 241–259. [Google Scholar] [CrossRef]
- Khaldan, A.; Bouamrane, S.; El Mchichi, R.E.M.L.; Maghat, H.; Lakhlifi, M.B.T.; Sbai, A. In search of new potent α-glucosidase inhibitors: Molecular docking and ADMET prediction. Mor. J. Chem. 2022, 10, 10–14. [Google Scholar] [CrossRef]
- Vajravijayan, S.; Nandhagopal, N.; Anantha Krishnan, D.; Gunasekaran, K. Isolation and characterization of an iridoid, Arbortristoside-C from Nyctanthes arbor-tristis Linn., a potential drug candidate for diabetes targeting α-glucosidase. J. Biomol. Struct. Dyn. 2022, 40, 337–347. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Peptides | A Parameter | MW [Da] | Protein ID | C_S | LOCK_S | BB_S | BAUER_S | C_M | LOCK_M | BB_M | BAUER_M |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | DIPPPPMDEK | 0.400 | 1137.54 | B5KJG2 | + | + | + | + | − | − | + | − |
| 2 | EAPPPPAEVH | 0.400 | 1042.51 | Q75NG9 | − | − | − | − | − | + | + | + |
| 3 | FDIPPPPMDE | 0.400 | 1156.51 | B5KJG2 | − | − | − | + | − | − | − | − |
| 4 | SFDIPPPPMD | 0.400 | 1114.50 | B5KJG2 | − | + | + | + | − | − | + | − |
| 5 | DLFPPPP | 0.429 | 781.40 | F1RYS7 | − | − | − | + | − | − | − | − |
| 6 | IIAPPER | 0.429 | 794.46 | B6VNT8; C7AI81; F1SLG5; I3LVD5; P68137; Q6QAQ1 | − | + | + | + | + | + | + | + |
| 7 | PPLIPPK | 0.429 | 760.48 | Q75ZZ6 | − | − | − | − | + | − | − | − |
| 8 | FDIPPPPMD | 0.444 | 1027.47 | B5KJG2 | − | − | − | + | − | − | − | − |
| 9 | IPPPPMDEK | 0.444 | 1022.51 | B5KJG2 | − | − | − | + | − | − | − | − |
| 10 | RPPPISPPP | 0.444 | 956.54 | F1RNQ0; I3LH78; I3LL74 | − | − | − | + | − | − | − | − |
| 11 | SFDIPPPPM | 0.444 | 999.47 | B5KJG2 | − | + | − | − | − | − | − | − |
| 12 | KSLRSGLLGDTLTEGGLSQLGRALREL | 0.476 | 2839.59 | F1RGE5 | + | − | − | − | − | − | − | − |
| 13 | VATPPPPPPPK | 0.546 | 1096.63 | I3LNG8 | − | − | + | − | − | − | − | − |
| 14 | DIPPPPM | 0.571 | 765.373 | B5KJG2 | − | − | − | + | − | − | − | − |
| 15 | TPPPPPPG | 0.625 | 758.40 | F1STN6 | − | − | − | + | − | − | − | − |
| 16 | TPPPPPPPK | 0.667 | 926.52 | I3LNG8 | − | − | − | + | − | − | − | − |
| Total number | 2 | 4 | 4 | 11 | 2 | 2 | 4 | 2 |
| No. Peptides | Allergencity 1 | A | D | M | E | T | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Caco-2 Permeability 2 | HIA 3 | PPB 4 | BBB 5 | VD 6 | Cyp450 2D6 7 [I/S] | T1/2 8 | LD50 9 | H-HT 10 | FDA 11 | |||
| 1 | DIPPPPMDEK | Probable Non-Allergen | −6.522 | 0.270 | 52.70 | 0.076 | −0.78 | 0.336/0.521 | 1.99 | 3.125 | 0.0 | 0.478 |
| 2 | EAPPPPAEVH | Probable Allergen | −6.576 | 0.220 | 51.77 | 0.178 | −0.74 | 0.376/0.473 | 1.92 | 3.226 | 0.0 | 0.31 |
| 3 | FDIPPPPMDE | Probable Non-Allergen | −6.526 | 0.284 | 61.95 | 0.119 | −0.87 | 0.407/0.488 | 1.95 | 3.157 | 0.0 | 0.346 |
| 4 | SFDIPPPPMD | Probable Non-Allergen | −3.223 | 0.262 | 60.42 | 0.052 | −0.85 | 0.375/0.479 | 1.93 | 3.178 | 0.0 | 0.308 |
| 5 | DLFPPPP | Probable Non-Allergen | −3.185 | 0.208 | 62.08 | 0.256 | −0.43 | 0.344/0.494 | 1.83 | 2.821 | 0.176 | 0.284 |
| 6 | IIAPPER | Probable Allergen | −6.383 | 0.288 | 49.89 | 0.105 | −0.52 | 0.359/0.526 | 1.68 | 2.838 | 0.056 | 0.418 |
| 7 | PPLIPPK | Probable Non-Allergen | −5.963 | 0.312 | 56.01 | 0.079 | −0.14 | 0.382/0.481 | 1.76 | 2.772 | 0.104 | 0.388 |
| 8 | FDIPPPPMD | Probable Non-Allergen | −6.511 | 0.284 | 58.38 | 0.110 | −0.80 | 0.384/0.499 | 1.88 | 3.143 | 0.0 | 0.324 |
| 9 | IPPPPMDEK | Probable Non-Allergen | −3.292 | 0.270 | 50.21 | 0.094 | −0.71 | 0.315/0.512 | 1.92 | 3.08 | 0.002 | 0.484 |
| 10 | RPPPISPPP | Probable Non-Allergen | −6.631 | 0.280 | 51.28 | 0.024 | −0.56 | 0.327/0.475 | 1.94 | 3.179 | 0.01 | 0.448 |
| 11 | SFDIPPPPM | Probable Non-Allergen | −6.483 | 0.262 | 58.47 | 0.052 | −0.76 | 0.387/0.478 | 1.90 | 3.102 | 0.0 | 0.306 |
| 12 | KSLRSGLLGDTLTEGGLSQLGRALREL | Probable Allergen | −6.221 | 0.161 | 59.80 | 0.041 | −0.25 | 0.445/0.437 | 2.15 | 3.239 | 0.0 | 0.44 |
| 13 | VATPPPPPPPK | Probable Non-Allergen | −6.352 | 0.197 | 50.76 | 0.084 | −0.30 | 0.343/0.518 | 2.08 | 3.314 | 0.0 | 0.428 |
| 14 | DIPPPPM | Probable Non-Allergen | −6.122 | 0.278 | 47.39 | 0.175 | −0.74 | 0.303/0.488 | 1.70 | 2.685 | 0.128 | 0.43 |
| 15 | TPPPPPPG | Probable Non-Allergen | −6.132 | 0.165 | 43.80 | 0.548 | −0.48 | 0.252/0.501 | 1.83 | 2.892 | 0.100 | 0.492 |
| 16 | TPPPPPPPK | Probable Non-Allergen | −6.246 | 0.173 | 46.57 | 0.137 | −0.34 | 0.270/0.548 | 1.99 | 3.218 | 0.024 | 0.472 |
| Cavity Number | ∆Gbinding [kcal/mol] | |||
|---|---|---|---|---|
| VATPPPPPPPK | DIPPPPM | TPPPPPPG | TPPPPPPPK | |
| 1 | −4.7 | −5.7 | −6.7 | −6.8 |
| 2 | −6.6 | −6.7 | −7.0 | −8.0 |
| 3 | −4.2 | −6.6 | −6.9 | −6.9 |
| 4 | −4.3 | −4.8 | −5.7 | −6.3 |
| 5 | −5.8 | −6.2 | −6.7 | −6.8 |
| 6 | −5.3 | −5.1 | −6.7 | −6.1 |
| 7 | −2.2 | −5.4 | −5.5 | −5.3 |
| 8 | −4.8 | −5.4 | −5.8 | −4.9 |
| 9 | 5.0 | −5.8 | −6.1 | −5.5 |
| 10 | 36.9 | −1.9 | −1.4 | −1.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kęska, P.; Stadnik, J.; Łupawka, A.; Michalska, A. Novel α-Glucosidase Inhibitory Peptides Identified In Silico from Dry-Cured Pork Loins with Probiotics through Peptidomic and Molecular Docking Analysis. Nutrients 2023, 15, 3539. https://doi.org/10.3390/nu15163539
Kęska P, Stadnik J, Łupawka A, Michalska A. Novel α-Glucosidase Inhibitory Peptides Identified In Silico from Dry-Cured Pork Loins with Probiotics through Peptidomic and Molecular Docking Analysis. Nutrients. 2023; 15(16):3539. https://doi.org/10.3390/nu15163539
Chicago/Turabian StyleKęska, Paulina, Joanna Stadnik, Aleksandra Łupawka, and Agata Michalska. 2023. "Novel α-Glucosidase Inhibitory Peptides Identified In Silico from Dry-Cured Pork Loins with Probiotics through Peptidomic and Molecular Docking Analysis" Nutrients 15, no. 16: 3539. https://doi.org/10.3390/nu15163539