
Nicotinamide Prevents Diabetic Brain Inflammation via NAD+-Dependent Deacetylation Mechanisms
 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
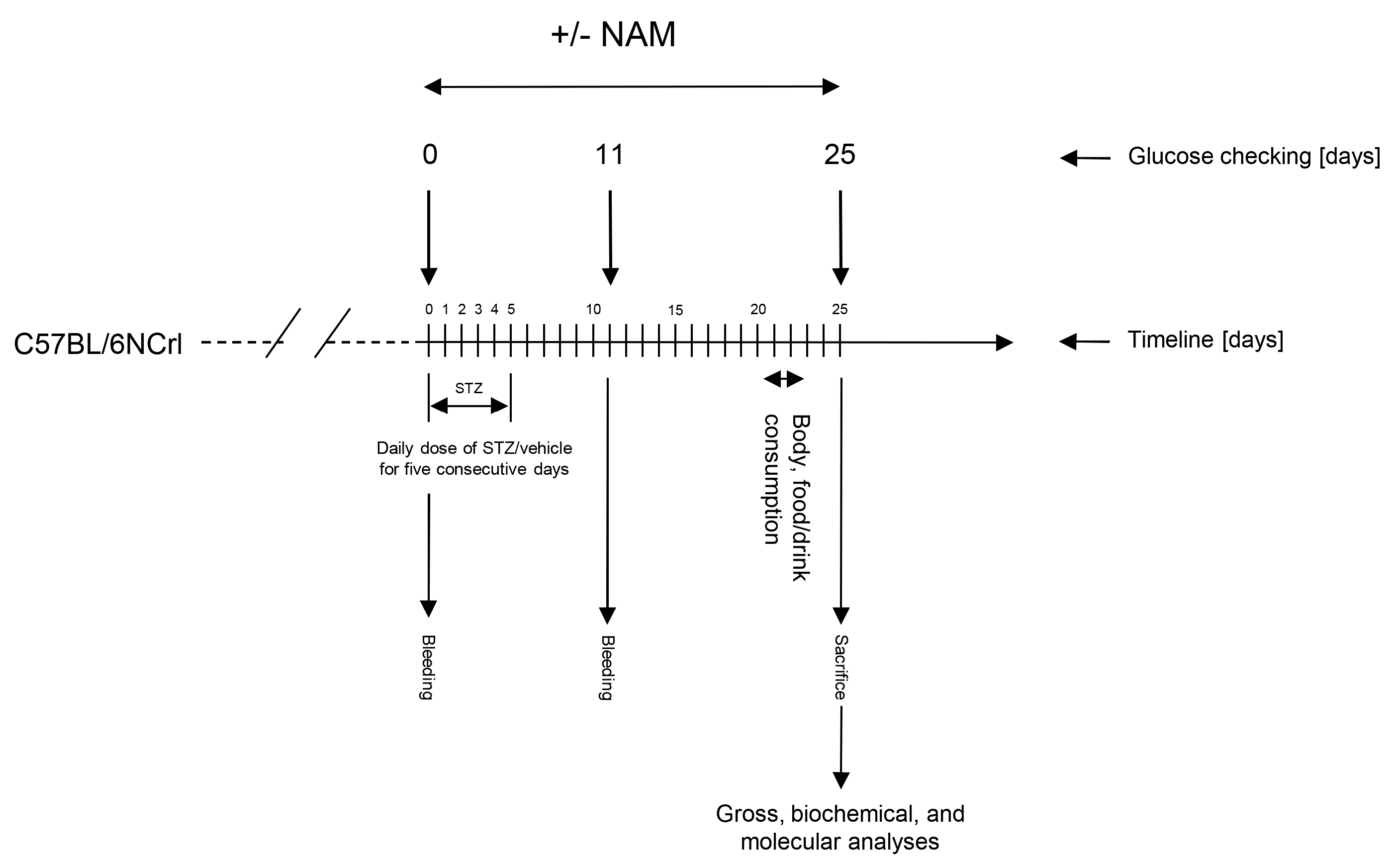
2.1. Mice and Treatments
2.2. Assessment of NAM on Diabetic Brain Inflammation Induced with STZ
2.3. Laboratory Analysis
2.4. Immunohistochemistry Analysis
2.5. Western Blotting
2.6. Quantitative Real-Time RT-PCR Analysis
2.7. Statistical Analysis
3. Results
3.1. NAM Effects on Gross and Biochemical Parameters of Diabetic Mice
3.2. NAM Attenuated NFκB Signaling and Microglial Activation in Diabetic Mice
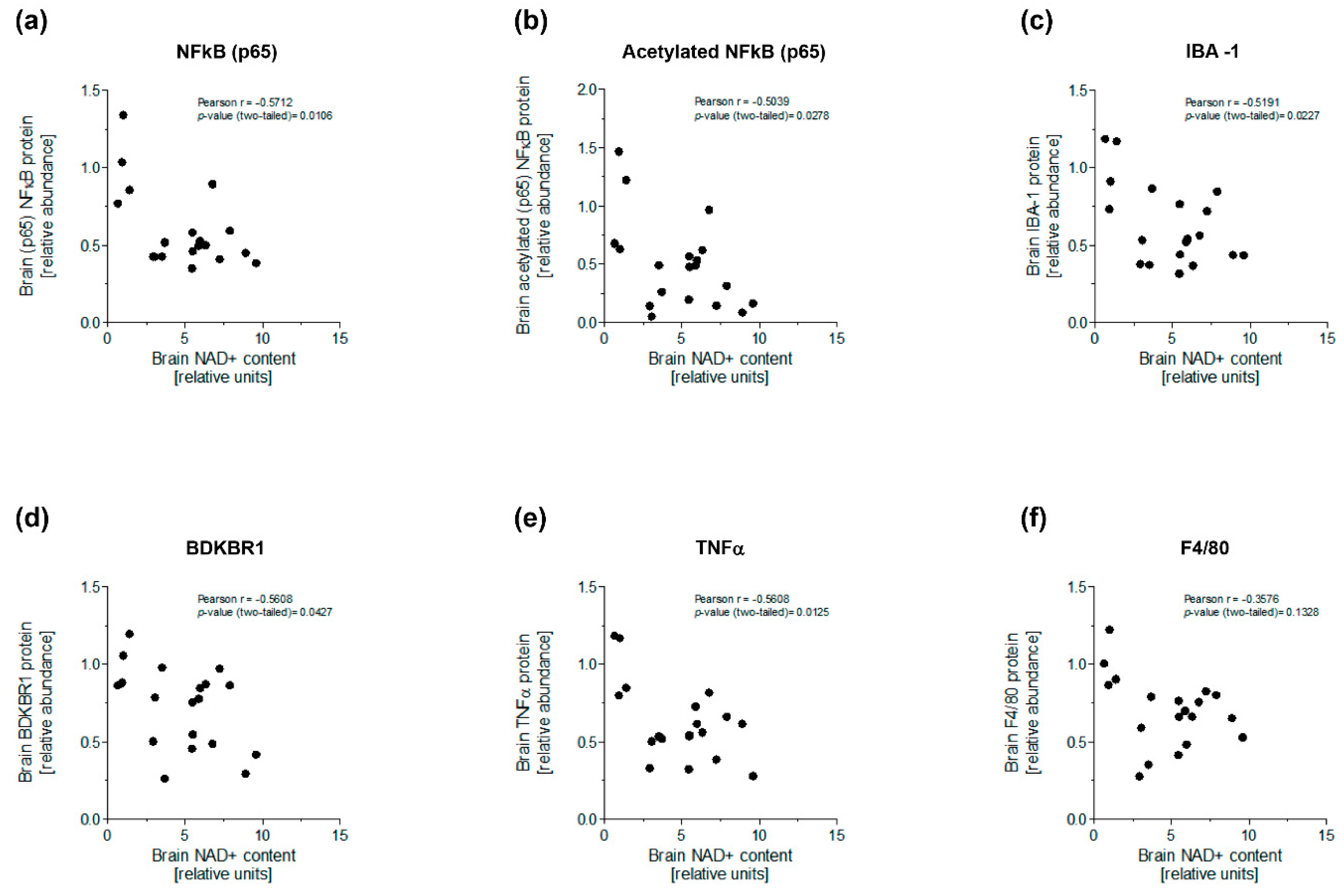
3.3. NAM Enhanced NAD+-Dependent Deacetylation of NFκB to Ameliorate Brain Inflammation of Diabetic Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Kappelle, A.C.; Bravenboer, B.; Erkelens, D.W.; Gispen, W.H. Cerebral function in diabetes mellitus. Diabetologia 1994, 37, 643–650. [Google Scholar] [CrossRef]
- Hamed, S.A. Brain injury with diabetes mellitus: Evidence, mechanisms and treatment implications. Expert. Rev. Clin. Pharmacol. 2017, 10, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Despa, F. Contributing Factors to Diabetic Brain Injury and Cognitive Decline. Diabetes Metab. J. 2019, 43, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Liu, T. Inflammatory cause of metabolic syndrome via brain stress and NF-kappaB. Aging 2012, 4, 98–115. [Google Scholar] [CrossRef]
- Feldman, E.L.; Nave, K.A.; Jensen, T.S.; Bennett, D.L.H. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 2017, 93, 1296–1313. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Santani, D. Role of NF-kappa B in the pathogenesis of diabetes and its associated complications. Pharmacol. Rep. 2009, 61, 595–603. [Google Scholar] [CrossRef]
- Israel, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef] [Green Version]
- Meffert, M.K.; Baltimore, D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005, 28, 37–43. [Google Scholar] [CrossRef]
- Chojdak-Lukasiewicz, J.; Bizon, A.; Waliszewska-Prosol, M.; Piwowar, A.; Budrewicz, S.; Pokryszko-Dragan, A. Role of Sirtuins in Physiology and Diseases of the Central Nervous System. Biomedicines 2022, 10, 2434. [Google Scholar] [CrossRef] [PubMed]
- Fricker, R.A.; Green, E.L.; Jenkins, S.I.; Griffin, S.M. The Influence of Nicotinamide on Health and Disease in the Central Nervous System. Int. J. Tryptophan Res. 2018, 11, 1178646918776658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.J. Pathogenesis of chronic hyperglycemia: From reductive stress to oxidative stress. J. Diabetes Res. 2014, 2014, 137919. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Szabo, C. Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: Endothelial dysfunction, as a common underlying theme. Antioxid. Redox Signal 2005, 7, 1568–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-kappaB activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.C.; Kong, Y.Y.; Li, G.Q.; Guan, Y.F.; Wang, P.; Miao, C.Y. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci. Rep. 2017, 7, 717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, U.; Evans, J.E.; Pearson, A.; Saltiel, N.; Cseresznye, A.; Darcey, T.; Ojo, J.; Keegan, A.P.; Oberlin, S.; Mouzon, B.; et al. Targeting sirtuin activity with nicotinamide riboside reduces neuroinflammation in a GWI mouse model. Neurotoxicology 2020, 79, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Yang, S.J. Supplementation with Nicotinamide Riboside Reduces Brain Inflammation and Improves Cognitive Function in Diabetic Mice. Int. J. Mol. Sci. 2019, 20, 4196. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef]
- de Castro, J.M.; Stein, D.J.; Medeiros, H.R.; de Oliveira, C.; Torres, I.L.S. Nicotinamide Riboside Neutralizes Hypothalamic Inflammation and Increases Weight Loss Without Altering Muscle Mass in Obese Rats Under Calorie Restriction: A Preliminary Investigation. Front. Nutr. 2021, 8, 648893. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD(+) in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef] [PubMed]
- Amjad, S.; Nisar, S.; Bhat, A.A.; Shah, A.R.; Frenneaux, M.P.; Fakhro, K.; Haris, M.; Reddy, R.; Patay, Z.; Baur, J.; et al. Role of NAD(+) in regulating cellular and metabolic signaling pathways. Mol. Metab. 2021, 49, 101195. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Kuan, Y.H.; Chen, H.Y.; Chen, T.Y.; Chen, S.T.; Huang, C.C.; Yang, I.P.; Hsu, Y.S.; Wu, T.S.; Lee, E.J. Intravenous administration of melatonin reduces the intracerebral cellular inflammatory response following transient focal cerebral ischemia in rats. J. Pineal Res. 2007, 42, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.C.; Holden, R.C.; Rasmussen, S.M.; Hoane, M.R.; Hylin, M.J. Effects of nicotinamide on spatial memory and inflammation after juvenile traumatic brain injury. Behav. Brain Res. 2019, 364, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Rehman, I.U.; Khan, A.; Ahmad, R.; Choe, K.; Park, H.Y.; Lee, H.J.; Atiq, A.; Park, J.; Hahm, J.R.; Kim, M.O. Neuroprotective Effects of Nicotinamide against MPTP-Induced Parkinson’s Disease in Mice: Impact on Oxidative Stress, Neuroinflammation, Nrf2/HO-1 and TLR4 Signaling Pathways. Biomedicines 2022, 10, 2929. [Google Scholar] [CrossRef]
- Mendez-Lara, K.A.; Letelier, N.; Farre, N.; Diarte-Anazco, E.M.G.; Nieto-Nicolau, N.; Rodriguez-Millan, E.; Santos, D.; Pallares, V.; Escola-Gil, J.C.; Vazquez Del Olmo, T.; et al. Nicotinamide Prevents Apolipoprotein B-Containing Lipoprotein Oxidation, Inflammation and Atherosclerosis in Apolipoprotein E-Deficient Mice. Antioxidants 2020, 9, 1162. [Google Scholar] [CrossRef]
- Mendez-Lara, K.A.; Rodriguez-Millan, E.; Sebastian, D.; Blanco-Soto, R.; Camacho, M.; Nan, M.N.; Diarte-Anazco, E.M.G.; Mato, E.; Lope-Piedrafita, S.; Roglans, N.; et al. Nicotinamide Protects Against Diet-Induced Body Weight Gain, Increases Energy Expenditure, and Induces White Adipose Tissue Beiging. Mol. Nutr. Food Res. 2021, 65, e2100111. [Google Scholar] [CrossRef]
- Crowe, A.R.; Yue, W. Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio Protoc. 2019, 9, e3465. [Google Scholar] [CrossRef]
- Spector, R. Niacinamide transport through the blood-brain barrier. Neurochem. Res. 1987, 12, 27–31. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Trammell, S.A.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.A.; Brenner, C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci. Rep. 2016, 6, 26933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lautrup, S.; Lou, G.; Aman, Y.; Nilsen, H.; Tao, J.; Fang, E.F. Microglial mitophagy mitigates neuroinflammation in Alzheimer’s disease. Neurochem. Int. 2019, 129, 104469. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The Role of Microglia and Macrophages in CNS Homeostasis, Autoimmunity, and Cancer. J. Immunol. Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuzawa, M.; Satoh, J.; Muto, G.; Muto, Y.; Nishimura, S.; Miyaguchi, S.; Qiang, X.L.; Toyota, T. Inhibitory effect of nicotinamide on in vitro and in vivo production of tumor necrosis factor-alpha. Immunol. Lett. 1997, 59, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, J.S.; Blomback, M.; Soderstrom, T. Nicotinamide is a potent inhibitor of proinflammatory cytokines. Clin. Exp. Immunol. 2003, 131, 48–52. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, Q.; Jiang, Y.; Liu, N.; Liu, Q.; Shi, F.D.; Hao, J.; Xu, Y.; Lo, E.H.; Wang, X. Augmented Brain Infiltration and Activation of Leukocytes After Cerebral Ischemia in Type 2 Diabetic Mice. Front. Immunol. 2019, 10, 2392. [Google Scholar] [CrossRef]
- Finger, C.E.; Moreno-Gonzalez, I.; Gutierrez, A.; Moruno-Manchon, J.F.; McCullough, L.D. Age-related immune alterations and cerebrovascular inflammation. Mol. Psychiatry 2022, 27, 803–818. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Marceau, F.; Bachvarov, D.R. Kinin receptors. Clin. Rev. Allergy Immunol. 1998, 16, 385–401. [Google Scholar] [CrossRef]
- Muriach, M.; Flores-Bellver, M.; Romero, F.J.; Barcia, J.M. Diabetes and the brain: Oxidative stress, inflammation, and autophagy. Oxid. Med. Cell Longev. 2014, 2014, 102158. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.C.; Hallows, W.C.; Denu, J.M. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal. Biochem. 2009, 394, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, C.; Lackie, T.; Williams, D.H.; Simone, P.S.; Zhang, Y.; Bloomer, R.J. Oral Administration of Nicotinamide Mononucleotide Increases Nicotinamide Adenine Dinucleotide Level in an Animal Brain. Nutrients 2022, 14, 300. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, K.; Choi, J.; Arvas, M.I.; Salimian, M.; Singh, S.; Xu, S.; Gullapalli, R.P.; Kristian, T.; Russell, J.W. Nicotinamide Mononucleotide Administration Prevents Experimental Diabetes-Induced Cognitive Impairment and Loss of Hippocampal Neurons. Int. J. Mol. Sci. 2020, 21, 3756. [Google Scholar] [CrossRef] [PubMed]
- Huynh, F.K.; Hershberger, K.A.; Hirschey, M.D. Targeting sirtuins for the treatment of diabetes. Diabetes Manag. 2013, 3, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gool, F.; Galli, M.; Gueydan, C.; Kruys, V.; Prevot, P.P.; Bedalov, A.; Mostoslavsky, R.; Alt, F.W.; De Smedt, T.; Leo, O. Intracellular NAD levels regulate tumor necrosis factor protein synthesis in a sirtuin-dependent manner. Nat. Med. 2009, 15, 206–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Cao, W.; Wang, Z.; Li, F.; Xiao, J.; Zeng, Q.; Wang, Y.; Li, S.; Ye, C.; Zheng, K. Nicotinamide n-Oxide Attenuates HSV-1-Induced Microglial Inflammation through Sirtuin-1/NF-kappaB Signaling. Int. J. Mol. Sci. 2022, 23, 16085. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Non-T1D | T1D | p-Value | ||
|---|---|---|---|---|---|
| Untreated | NAM LD | NAM HD | |||
| Gross | |||||
| Final body weight (g) | 29.16 ± 1.01 | 21.65 ± 1.26 * | 21.69 ± 0.97 * | 21.08 ± 0.67 * | <0.05 |
| Liver weight (g) | 1.22 ± 0.04 | 1.26 ± 0.07 | 1.17 ± 0.06 | 1.02 ± 0.08 | 0.2638 |
| Kidneys’ weight (g) | 0.38 ± 0.01 | 0.39 ± 0.01 | 0.38 ± 0.02 | 0.37 ± 0.02 | 0.4528 |
| Brain weight (g) | 0.40 ± 0.01 | 0.38 ± 0.00 | 0.39 ± 0.01 | 0.39 ± 0.01 | 0.9660 |
| Food intake (g/day) | 2.65 ± 0.20 | 7.11 ± 0.31 * | 6.77 ± 0.92 * | 7.52 ± 0.66 * | <0.05 |
| Water consumption (mL/day) | 4.73 ± 0.25 | 36.21 ± 2.41 * | 37.67 ± 1.21 * | 32.18 ± 1.57 * | <0.05 |
| NAM intake (g/day/kg) | -- | -- | 1.75 ± 0.10 | 3.73 ± 0.26 ‡ | <0.05 |
| Plasma biochemistry | |||||
| Glucose (mmol/L) | 12.3 ± 0.6 | 40.7 ± 2.3 * | 41.1 ± 2.3 * | 41.7 ± 3.0 * | <0.05 |
| Insulin (ng/mL) | 0.65 ± 0.06 | 0.12 ± 0.03 * | 0.26 ± 0.15 * | 0.15 ± 0.02 * | <0.0001 |
| AST (U/L) | 272.0 ± 142.4 | 405.6 ± 139.1 | 364.6 ± 228.6 | 504.2 ± 211.9 | 0.8676 |
| ALT (U/L) | 60.15 ± 33.02 | 156.6 ± 41.92 | 45.53 ± 29.74 | 53.72 ± 6.95 | 0.1898 |
| Total cholesterol (mmol/L) | 3.06 ± 0.32 | 2.95 ± 0.15 | 3.96 ± 0.73 | 3.08 ± 0.23 | 0.1661 |
| Triglycerides (mmol/L) | 0.60 ± 0.13 | 1.61 ± 0.23 * | 0.61 ± 0.10 † | 0.93 ± 0.18 | <0.01 |
| Brain NAD+ | |||||
| NAD+ (pmol/mg protein) | 1.86 ± 0.45 | 1.01 ± 0.22 a | 3.67 ± 0.47 † | 3.50 ± 0.16 † | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Méndez, J.K.; Niño-Narvión, J.; Martinez-Santos, P.; Diarte-Añazco, E.M.G.; Méndez-Lara, K.A.; del Olmo, T.V.; Rotllan, N.; Julián, M.T.; Alonso, N.; Mauricio, D.; et al. Nicotinamide Prevents Diabetic Brain Inflammation via NAD+-Dependent Deacetylation Mechanisms. Nutrients 2023, 15, 3083. https://doi.org/10.3390/nu15143083
Torres-Méndez JK, Niño-Narvión J, Martinez-Santos P, Diarte-Añazco EMG, Méndez-Lara KA, del Olmo TV, Rotllan N, Julián MT, Alonso N, Mauricio D, et al. Nicotinamide Prevents Diabetic Brain Inflammation via NAD+-Dependent Deacetylation Mechanisms. Nutrients. 2023; 15(14):3083. https://doi.org/10.3390/nu15143083
Chicago/Turabian StyleTorres-Méndez, Jeimy Katherine, Julia Niño-Narvión, Patricia Martinez-Santos, Elena María Goretti Diarte-Añazco, Karen Alejandra Méndez-Lara, Tania Vázquez del Olmo, Noemi Rotllan, Maria Teresa Julián, Núria Alonso, Didac Mauricio, and et al. 2023. "Nicotinamide Prevents Diabetic Brain Inflammation via NAD+-Dependent Deacetylation Mechanisms" Nutrients 15, no. 14: 3083. https://doi.org/10.3390/nu15143083