
Polygonum cuspidatum Extract (Pc-Ex) Containing Emodin Suppresses Lung Cancer-Induced Cachexia by Suppressing TCF4/TWIST1 Complex-Induced PTHrP Expression

Abstract
:1. Introduction
2. Materials and Methods
2.1. Quantitative Real-Time PCR (qRT-PCR)
2.2. Co-Immunoprecipitation and Western Blotting
2.3. Mammalian Two-Hybrid Luciferase Assay
2.4. Cell Culture and Transient Transfection
2.5. Adenoviral Transduction, Mammalian Expression Plasmids and Gene Cloning
2.6. Animal Experiment
2.7. ELISA for Measurement of PTHrP
2.8. Preparation of Pc-Ex and Measurement of Emodin Concentration
3. Results
3.1. PTHLH Expression Is Upregulated in Response to TGFβ1 in Lung Cancer
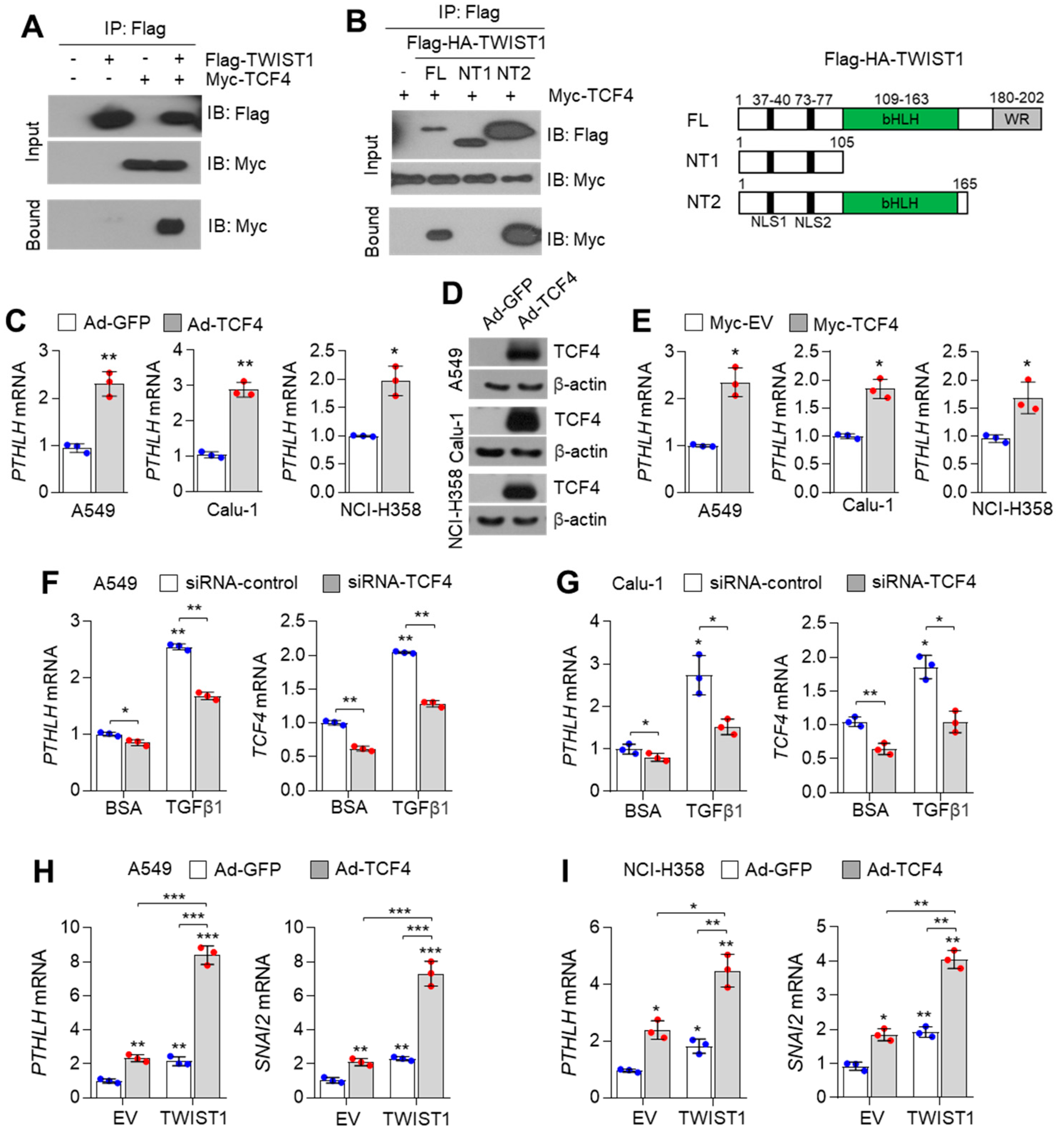
3.2. TCF4 Regulates PTHLH Expression in Lung Cancer Cells
3.3. Identification of a Natural Compound Targeting TCF4–TWIST1 Interaction Using Mammalian Two-Hybrid System-Based High-Throughput Screening (HTS)
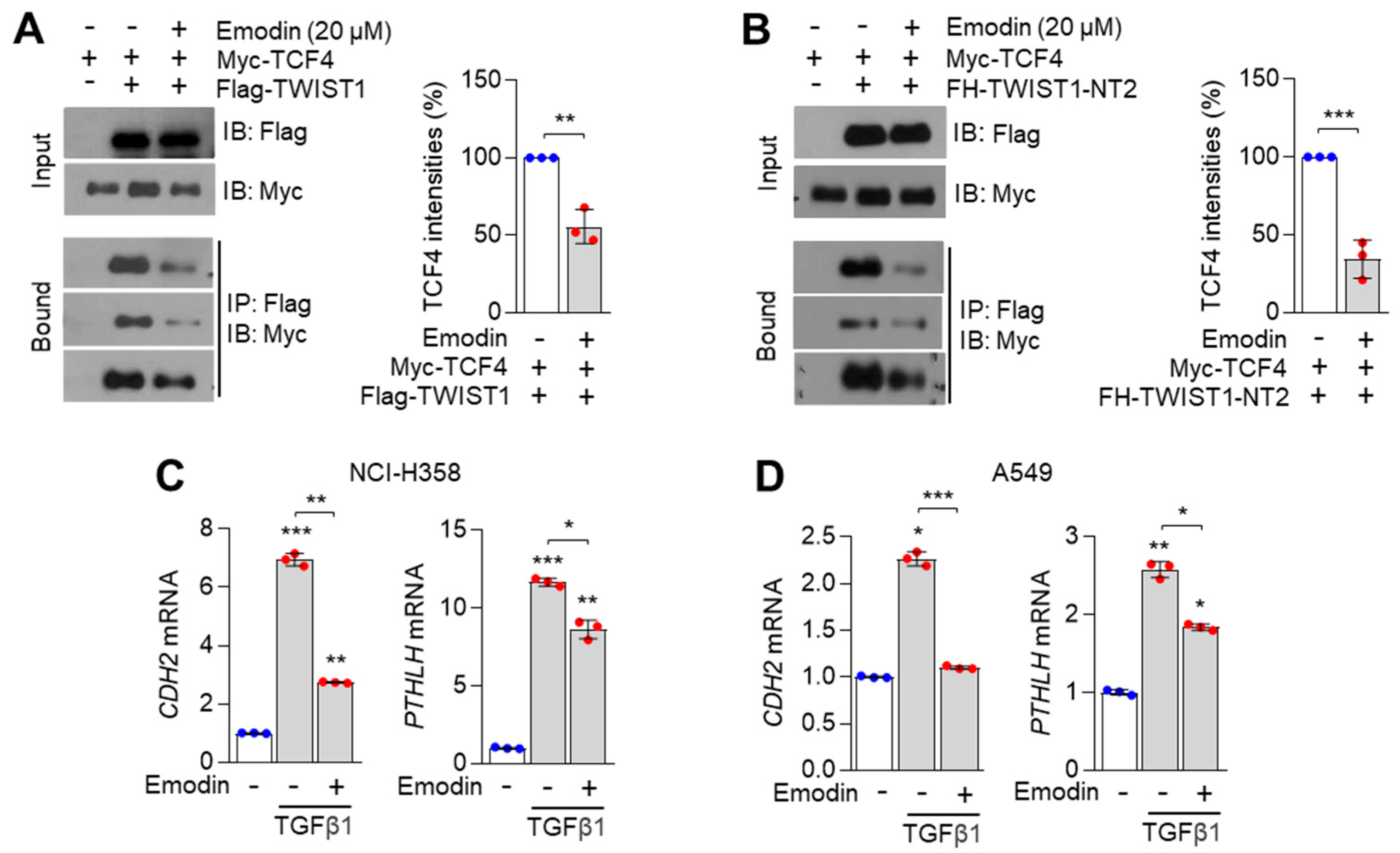
3.4. Emodin Inhibits TCF4/TWIST1 Complex-Induced PTHLH Expression
3.5. Emodin-Enriched Polygonum cuspidatum Extract (Pc-Ex) Inhibits TCF4–TWIST1 Interaction and Consequently Suppresses TGFβ1-Induced PTHLH Expression
3.6. Pc-Ex Attenuates Lung Cancer-Induced Cachexia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, R.; Liu, Z.; Jiao, R.; Zhang, C.; Yu, Q.; Han, S.; Duan, Z. Updates on the pathogenesis of advanced lung cancer-induced cachexia. Thorac. Cancer 2019, 10, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Hanna, D.L.; Zhang, W.; Baba, H.; Lenz, H.J. Molecular Pathways: Cachexia Signaling-A Targeted Approach to Cancer Treatment. Clin. Cancer Res. 2016, 22, 3999–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kir, S.; White, J.P.; Kleiner, S.; Kazak, L.; Cohen, P.; Baracos, V.E.; Spiegelman, B.M. Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 2014, 513, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Matthys, P.; Billiau, A. Cytokines and cachexia. Nutrition 1997, 13, 763–770. [Google Scholar] [CrossRef]
- Ma, J.F.; Sanchez, B.J.; Hall, D.T.; Tremblay, A.K.; Di Marco, S.; Gallouzi, I.E. STAT3 promotes IFNgamma/TNFalpha-induced muscle wasting in an NF-kappaB-dependent and IL-6-independent manner. EMBO Mol. Med. 2017, 9, 622–637. [Google Scholar] [CrossRef]
- Guttridge, D.C. A TGF-beta pathway associated with cancer cachexia. Nat. Med. 2015, 21, 1248–1249. [Google Scholar] [CrossRef] [Green Version]
- Lima, J.; Simoes, E.; de Castro, G.; Morais, M.; de Matos-Neto, E.M.; Alves, M.J.; Pinto, N.I.; Figueredo, R.G.; Zorn, T.M.T.; Felipe-Silva, A.S.; et al. Tumour-derived transforming growth factor-beta signalling contributes to fibrosis in patients with cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 10, 1045–1059. [Google Scholar] [CrossRef]
- Daas, S.I.; Rizeq, B.R.; Nasrallah, G.K. Adipose tissue dysfunction in cancer cachexia. J. Cell. Physiol. 2018, 234, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Kir, S.; Spiegelman, B.M. Cachexia & Brown Fat: A Burning Issue in Cancer. Trends Cancer 2016, 2, 461–463. [Google Scholar] [CrossRef] [Green Version]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berardi, E.; Madaro, L.; Lozanoska-Ochser, B.; Adamo, S.; Thorrez, L.; Bouche, M.; Coletti, D. A Pound of Flesh: What Cachexia Is and What It Is Not. Diagnostics 2021, 11, 116. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, T.M.; Golzarri-Arroyo, L.; Pin, F.; Barreto, R.; Dickinson, S.L.; Couch, M.E.; Bonetto, A. Metabolic Biomarkers for the Early Detection of Cancer Cachexia. Front. Cell Dev. Biol. 2021, 9, 720096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Li, J.; Assaker, G.; Camirand, A.; Sabri, S.; Karaplis, A.C.; Kremer, R. Parathyroid Hormone-Related Protein (PTHrP): An Emerging Target in Cancer Progression and Metastasis. Adv. Exp. Med. Biol. 2019, 1164, 161–178. [Google Scholar] [CrossRef]
- Miki, T.; Yano, S.; Hanibuchi, M.; Sone, S. Bone metastasis model with multiorgan dissemination of human small-cell lung cancer (SBC-5) cells in natural killer cell-depleted SCID mice. Oncol. Res. 2000, 12, 209–217. [Google Scholar] [CrossRef]
- Oh, T.I.; Lee, M.; Lee, Y.M.; Kim, G.H.; Lee, D.; You, J.S.; Kim, S.H.; Choi, M.; Jang, H.; Park, Y.M.; et al. PGC1alpha Loss Promotes Lung Cancer Metastasis through Epithelial-Mesenchymal Transition. Cancers 2021, 13, 1772. [Google Scholar] [CrossRef]
- Wang, Y.; Lei, R.; Zhuang, X.; Zhang, N.; Pan, H.; Li, G.; Hu, J.; Pan, X.; Tao, Q.; Fu, D.; et al. DLC1-dependent parathyroid hormone-like hormone inhibition suppresses breast cancer bone metastasis. J. Clin. Investig. 2014, 124, 1646–1659. [Google Scholar] [CrossRef] [Green Version]
- Calvo, N.; Carriere, P.; Martin, M.J.; Gigola, G.; Gentili, C. PTHrP treatment of colon cancer cells promotes tumor associated-angiogenesis by the effect of VEGF. Mol. Cell Endocrinol. 2019, 483, 50–63. [Google Scholar] [CrossRef]
- Pitarresi, J.R.; Norgard, R.J.; Chiarella, A.M.; Suzuki, K.; Bakir, B.; Sahu, V.; Li, J.; Zhao, J.; Marchand, B.; Wengyn, M.D.; et al. PTHrP Drives Pancreatic Cancer Growth and Metastasis and Reveals a New Therapeutic Vulnerability. Cancer Discov. 2021, 11, 1774–1791. [Google Scholar] [CrossRef]
- Downs, T.M.; Burton, D.W.; Araiza, F.L.; Hastings, R.H.; Deftos, L.J. PTHrP stimulates prostate cancer cell growth and upregulates aldo-keto reductase 1C3. Cancer Lett. 2011, 306, 52–59. [Google Scholar] [CrossRef]
- Forrest, M.P.; Hill, M.J.; Quantock, A.J.; Martin-Rendon, E.; Blake, D.J. The emerging roles of TCF4 in disease and development. Trends Mol. Med. 2014, 20, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.H.; Goo, B.K.; Moon, J.; Choi, Y.; Hwang, J.J.; Kim, C.S.; Bae, K.S.; Choi, J.; Cho, S.Y.; Yang, S.H.; et al. Induction of immunoglobulin transcription factor 2 and resistance to MEK inhibitor in melanoma cells. Oncotarget 2017, 8, 41387–41400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appaiah, H.; Bhat-Nakshatri, P.; Mehta, R.; Thorat, M.; Badve, S.; Nakshatri, H. ITF2 is a target of CXCR4 in MDA-MB-231 breast cancer cells and is associated with reduced survival in estrogen receptor-negative breast cancer. Cancer Biol. Ther. 2010, 10, 600–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobrado, V.R.; Moreno-Bueno, G.; Cubillo, E.; Holt, L.J.; Nieto, M.A.; Portillo, F.; Cano, A. The class I bHLH factors E2-2A and E2-2B regulate EMT. J. Cell Sci. 2009, 122, 1014–1024. [Google Scholar] [CrossRef] [Green Version]
- Kolligs, F.T.; Nieman, M.T.; Winer, I.; Hu, G.; Van Mater, D.; Feng, Y.; Smith, I.M.; Wu, R.; Zhai, Y.; Cho, K.R.; et al. ITF-2, a downstream target of the Wnt/TCF pathway, is activated in human cancers with beta-catenin defects and promotes neoplastic transformation. Cancer Cell 2002, 1, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Lim, J.H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.R.; Puigserver, P. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016, 537, 422–426. [Google Scholar] [CrossRef] [Green Version]
- Pernia, O.; Sastre-Perona, A.; Rodriguez-Antolin, C.; Garcia-Guede, A.; Palomares-Bralo, M.; Rosas, R.; Sanchez-Cabrero, D.; Cruz, P.; Rodriguez, C.; Diestro, M.; et al. A Novel Role for the Tumor Suppressor Gene ITF2 in Tumorigenesis and Chemotherapy Response. Cancers 2020, 12, 786. [Google Scholar] [CrossRef] [Green Version]
- Brandl, L.; Horst, D.; de Toni, E.; Kirchner, T.; Herbst, A.; Kolligs, F.T. ITF-2B protein levels are correlated with favorable prognosis in patients with colorectal carcinomas. Am. J. Cancer Res. 2015, 5, 2241–2248. [Google Scholar]
- Grill, J.I.; Herbst, A.; Brandl, L.; Kong, L.; Schneider, M.R.; Kirchner, T.; Wolf, E.; Kolligs, F.T. Inactivation of Itf2 promotes intestinal tumorigenesis in Apc(Min/+) mice. Biochem. Biophys. Res. Commun. 2015, 461, 249–253. [Google Scholar] [CrossRef]
- Shin, H.W.; Choi, H.; So, D.; Kim, Y.I.; Cho, K.; Chung, H.J.; Lee, K.H.; Chun, Y.S.; Cho, C.H.; Kang, G.H.; et al. ITF2 prevents activation of the beta-catenin-TCF4 complex in colon cancer cells and levels decrease with tumor progression. Gastroenterology 2014, 147, 430–442.e8. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Waardenberg, A.J.; Demuth, M.; Osteil, P.; Sun, J.Q.J.; Loebel, D.A.F.; Graham, M.; Tam, P.P.L.; Fossat, N. TWIST1 Homodimers and Heterodimers Orchestrate Lineage-Specific Differentiation. Mol. Cell. Biol. 2020, 40, e00663-19. [Google Scholar] [CrossRef] [PubMed]
- Marwitz, S.; Depner, S.; Dvornikov, D.; Merkle, R.; Szczygiel, M.; Muller-Decker, K.; Lucarelli, P.; Wasch, M.; Mairbaurl, H.; Rabe, K.F.; et al. Downregulation of the TGFbeta Pseudoreceptor BAMBI in Non-Small Cell Lung Cancer Enhances TGFbeta Signaling and Invasion. Cancer Res. 2016, 76, 3785–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, H.W.; Lee, S.S.; Chang, C.Y.; Lang, Y.D.; Jou, Y.S. A New Switch for TGFbeta in Cancer. Cancer Res. 2019, 79, 3797–3805. [Google Scholar] [CrossRef] [Green Version]
- Gordian, E.; Welsh, E.A.; Gimbrone, N.; Siegel, E.M.; Shibata, D.; Creelan, B.C.; Cress, W.D.; Eschrich, S.A.; Haura, E.B.; Munoz-Antonia, T. Transforming growth factor beta-induced epithelial-to-mesenchymal signature predicts metastasis-free survival in non-small cell lung cancer. Oncotarget 2019, 10, 810–824. [Google Scholar] [CrossRef] [Green Version]
- Kakonen, S.M.; Selander, K.S.; Chirgwin, J.M.; Yin, J.J.; Burns, S.; Rankin, W.A.; Grubbs, B.G.; Dallas, M.; Cui, Y.; Guise, T.A. Transforming growth factor-beta stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. J. Biol. Chem. 2002, 277, 24571–24578. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, R.K.; Ballschmieter, P.; Nordheim, A.; Dittmer, J. Transforming growth factor beta regulates parathyroid hormone-related protein expression in MDA-MB-231 breast cancer cells through a novel Smad/Ets synergism. J. Biol. Chem. 2001, 276, 46661–46670. [Google Scholar] [CrossRef] [Green Version]
- Yasui, T.; Uemura, H.; Irahara, M.; Aono, T. Effects of transforming growth factor-beta on the production of parathyroid hormone-related peptide in a human ovarian cancer cell line in vitro. J. Obstet. Gynaecol. Res. 1997, 23, 231–238. [Google Scholar] [CrossRef]
- Schacter, L.; Rozencweig, M.; Canetta, R.; Kelley, S.; Nicaise, C.; Smaldone, L. Megestrol acetate: Clinical experience. Cancer Treat. Rev. 1989, 16, 49–63. [Google Scholar] [CrossRef]
- Cha, T.L.; Qiu, L.; Chen, C.T.; Wen, Y.; Hung, M.C. Emodin down-regulates androgen receptor and inhibits prostate cancer cell growth. Cancer Res. 2005, 65, 2287–2295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun-Guang, W.; Jun-Qing, Y.; Bei-Zhong, L.; Dan-Ting, J.; Chong, W.; Liang, Z.; Dan, Z.; Yan, W. Anti-tumor activity of emodin against human chronic myelocytic leukemia K562 cell lines in vitro and in vivo. Eur. J. Pharmacol. 2010, 627, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.Y.; Song, G.P.; Deng, J.P.; Jiang, L.Z.; Xiong, P.; Yang, B.J.; Liu, S.S. Antitumor Effects and Mechanism of Novel Emodin Rhamnoside Derivatives against Human Cancer Cells In Vitro. PLoS ONE 2015, 10, e0144781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Liao, Y.; He, S.; Shi, J.; Peng, L.; Xu, X.; Xie, F.; Diao, N.; Huang, J.; Xie, Q.; et al. Autocrine parathyroid hormone-like hormone promotes intrahepatic cholangiocarcinoma cell proliferation via increased ERK/JNK-ATF2-cyclinD1 signaling. J. Transl. Med. 2017, 15, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.M.; Lin, Y.F.; Su, C.Y.; Peng, H.Y.; Chang, Y.C.; Hsiao, J.R.; Chen, C.L.; Chang, J.Y.; Shieh, Y.S.; Hsiao, M.; et al. Parathyroid Hormone-Like Hormone is a Poor Prognosis Marker of Head and Neck Cancer and Promotes Cell Growth via RUNX2 Regulation. Sci. Rep. 2017, 7, 41131. [Google Scholar] [CrossRef] [Green Version]
- Walia, M.K.; Ho, P.M.; Taylor, S.; Ng, A.J.; Gupte, A.; Chalk, A.M.; Zannettino, A.C.; Martin, T.J.; Walkley, C.R. Activation of PTHrP-cAMP-CREB1 signaling following p53 loss is essential for osteosarcoma initiation and maintenance. Elife 2016, 5, e13446. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wang, Z.; Cui, R.; He, H.; Lin, X.; Sheng, Y.; Zhang, H. Co-expression of parathyroid hormone related protein and TGF-beta in breast cancer predicts poor survival outcome. BMC Cancer 2015, 15, 925. [Google Scholar] [CrossRef] [Green Version]
- Padua, D.; Massague, J. Roles of TGFbeta in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Mologni, L.; Dekhil, H.; Ceccon, M.; Purgante, S.; Lan, C.; Cleris, L.; Magistroni, V.; Formelli, F.; Gambacorti-Passerini, C.B. Colorectal tumors are effectively eradicated by combined inhibition of {beta}-catenin, KRAS, and the oncogenic transcription factor ITF2. Cancer Res. 2010, 70, 7253–7263. [Google Scholar] [CrossRef] [Green Version]
- Herbst, A.; Bommer, G.T.; Kriegl, L.; Jung, A.; Behrens, A.; Csanadi, E.; Gerhard, M.; Bolz, C.; Riesenberg, R.; Zimmermann, W.; et al. ITF-2 is disrupted via allelic loss of chromosome 18q21, and ITF-2B expression is lost at the adenoma-carcinoma transition. Gastroenterology 2009, 137, 639–648.e9. [Google Scholar] [CrossRef] [PubMed]
- Vicent, S.; Luis-Ravelo, D.; Anton, I.; Garcia-Tunon, I.; Borras-Cuesta, F.; Dotor, J.; De Las Rivas, J.; Lecanda, F. A novel lung cancer signature mediates metastatic bone colonization by a dual mechanism. Cancer Res. 2008, 68, 2275–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flora, A.; Garcia, J.J.; Thaller, C.; Zoghbi, H.Y. The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc. Natl. Acad. Sci. USA 2007, 104, 15382–15387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Qin, R.; Li, X.; Zhou, H. Botany, phytochemistry, pharmacology, and potential application of Polygonum cuspidatum Sieb.et Zucc.: A review. J. Ethnopharmacol. 2013, 148, 729–745. [Google Scholar] [CrossRef]
- Dong, X.; Fu, J.; Yin, X.; Cao, S.; Li, X.; Lin, L.; Huyiligeqi; Ni, J. Emodin: A Review of its Pharmacology, Toxicity and Pharmacokinetics. Phytother. Res. 2016, 30, 1207–1218. [Google Scholar] [CrossRef]
- Monisha, B.A.; Kumar, N.; Tiku, A.B. Emodin and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 47–73. [Google Scholar] [CrossRef]
- Sougiannis, A.T.; Enos, R.T.; VanderVeen, B.N.; Velazquez, K.T.; Kelly, B.; McDonald, S.; Cotham, W.; Chatzistamou, I.; Nagarkatti, M.; Fan, D.; et al. Safety of natural anthraquinone emodin: An assessment in mice. BMC Pharmacol. Toxicol. 2021, 22, 9. [Google Scholar] [CrossRef]
- Hu, L.; Cui, R.; Liu, H.; Wang, F. Emodin and rhein decrease levels of hypoxia-inducible factor-1alpha in human pancreatic cancer cells and attenuate cancer cachexia in athymic mice carrying these cells. Oncotarget 2017, 8, 88008–88020. [Google Scholar] [CrossRef]
- Liu, Q.; Hodge, J.; Wang, J.; Wang, Y.; Wang, L.; Singh, U.; Li, Y.; Yao, Y.; Wang, D.; Ai, W.; et al. Emodin reduces Breast Cancer Lung Metastasis by suppressing Macrophage-induced Breast Cancer Cell Epithelial-mesenchymal transition and Cancer Stem Cell formation. Theranostics 2020, 10, 8365–8381. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, J.; Qian, J.; Wu, G.; Ma, Z. Emodin alleviates CCl4induced liver fibrosis by suppressing epithelialmesenchymal transition and transforming growth factorbeta1 in rats. Mol. Med. Rep. 2018, 18, 3262–3270. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Li, H.; Zhang, S.; Xiong, X.; Chen, K.; Jiang, P.; Jiang, K.; Deng, G. Emodin ameliorates renal fibrosis in rats via TGF-beta1/Smad signaling pathway and function study of Smurf 2. Int. Urol. Nephrol. 2018, 50, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Chen, R.; Cao, Y.; Wang, Y.; Song, K.; Zhang, Y.; Yang, J. Emodin suppresses TGF-beta1-induced epithelial-mesenchymal transition in alveolar epithelial cells through Notch signaling pathway. Toxicol. Appl. Pharmacol. 2017, 318, 11998–12007. [Google Scholar] [CrossRef] [PubMed]
- Urosevic, J.; Garcia-Albeniz, X.; Planet, E.; Real, S.; Cespedes, M.V.; Guiu, M.; Fernandez, E.; Bellmunt, A.; Gawrzak, S.; Pavlovic, M.; et al. Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat. Cell Biol. 2014, 16, 685–694. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequences (5′-3′) | Reverse Sequences (5′-3′) |
|---|---|---|
| CDH2 (h) | CCACCTTAAAATCTGCAGGC | GTGCATGAAGGACAGCCTCT |
| SNAI2 (h) | TGACCTGTCTGCAAATGCTC | CAGACCCTGGTTGCTTCAA |
| TCF4 (h) | CATAGGGAGTCCCATCTCCA | GGACCAACTTCTTTGGCAAG |
| PTHLH (h) | TTGTCATGGAGGAGCTGATG | CGGTGTTCCTGCTGAGCTAC |
| 36B4 (h) | TGGTGATACCTAAAGCCTGGAA | CATGTTGCTGGCCAATAAGG |
| Mstn (m) | CAGGAGAAGATGGGCTGAATC | AGTGCTCATCGCAGTCAAG |
| Fbxo32 (m) | ACCCAAGAAGAGAGCAGTATG | GACTCCCAGCCATCCAATTA |
| Trim63 (m) | CAAGGAACACGAAGACGAGAA | TCCTCCAGCTGAGAGATGAT |
| IGF1 (m) | GTCGTCTTCACACCTCTTCTAC | CTCATCCACAATGCCTGTCT |
| UCP1 (m) | ACAGAAGGATTGCCGAAACT | CTGTAGGCTGCCCAATGAA |
| PGC1α (m) | AAACTGACTTCGAGCTGTACTT | CCCATGAGGTATTGACCATCTC |
| Acox1 (m) | TGCCTTTGTTGTCCCTATCC | GTCCATCTTCAGGTAGCCATTAT |
| Glut1 (m) | TCTGTCGGCCTCTTTGTTAATC | CCAGTTTGGAGAAGCCCATAA |
| B-actin (m) | ACGAGGCCCAGAGCAAGAG | TCTCCAAGTCGTCCCAGTTG |
| Constructs | Sequences (5′-3′) |
|---|---|
| Flag-HA-TWIST1-FL | [F] CCGCTCGAGATGATGCAGGACGTG |
| [R] CCGGAATTCTTAGTGGGACGCGGACAT | |
| Flag-HA-TWIST1-NT1 | [F] CCGCTCGAGATGATGCAGGACGTG |
| [R] CCGGAATTCTTACTCCTCGTAAGACTG | |
| Flag-HA-TWIST1-NT2 | [F] CCGCTCGAGATGATGCAGGACGTG |
| [R] CCGGAATTCTTAGCTCTGGAGGACCTG | |
| pFN10A-TCF4-FL | [F] ATAGGCGATCGCCATGCATCACCAACAGCGA |
| [R] AGCTTTGTTTAAACCATCTGTCCCATGTGATTCGATGC | |
| pFN10A-TCF4-N | [F] AGCTTTGTTTAAACTCCTGGTGGCATGCCTCT |
| [R] AGCTTTGTTTAAACCTGTGGAATATGAGAAGAGTTGCCC | |
| pFN10A-TCF4-M | [F] ATAGGCGATCGCCATGTCCAGCAGCTACTGTAGCC |
| [R] AGCTTTGTTTAAACTCCTGGTGGCATGCCTCT | |
| pFN10A-TCF4-C | [F] ATAGGCGATCGCCATGCTACAGGGGCAGAGTGT |
| [R] AGCTTTGTTTAAACCATCTGTCCCATGTGATTCGATGC | |
| pFN11A-TWIST1 | [F] ATAGGCGATCGCCATGATGCAGGACGTGTCCA |
| [R] AGCTTTGTTTAAACGTGGGACGCGGACATGGA |
| Nutrition | AIN93G (g) | 2% Pc-Ex (g) |
|---|---|---|
| Casein | 200 | 194.38 |
| Corn starch | 397.486 | 383.34 |
| Dextrose | 132 | 132 |
| Sucrose | 100 | 100 |
| Cellulose | 50 | 50 |
| Soybean Oil | 70 | 69.76 |
| t-Butylhydroquinone | 0.014 | 0.014 |
| Salt Mix | 35 | 35 |
| Vitamin Mix | 10 | 10 |
| L-cystine | 3 | 3 |
| Choline Bitartrate | 2.5 | 2.5 |
| Pc-Ex | 0 | 20 |
| Total | 1000 | 1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, X.-Q.; Kim, Y.-S.; Lee, Y.-M.; Lee, M.; Lim, W.-J.; Yim, W.-J.; Han, M.-W.; Lim, J.-H. Polygonum cuspidatum Extract (Pc-Ex) Containing Emodin Suppresses Lung Cancer-Induced Cachexia by Suppressing TCF4/TWIST1 Complex-Induced PTHrP Expression. Nutrients 2022, 14, 1508. https://doi.org/10.3390/nu14071508
Fang X-Q, Kim Y-S, Lee Y-M, Lee M, Lim W-J, Yim W-J, Han M-W, Lim J-H. Polygonum cuspidatum Extract (Pc-Ex) Containing Emodin Suppresses Lung Cancer-Induced Cachexia by Suppressing TCF4/TWIST1 Complex-Induced PTHrP Expression. Nutrients. 2022; 14(7):1508. https://doi.org/10.3390/nu14071508
Chicago/Turabian StyleFang, Xue-Quan, Young-Seon Kim, Yoon-Mi Lee, Mingyu Lee, Woo-Jin Lim, Woo-Jong Yim, Min-Woo Han, and Ji-Hong Lim. 2022. "Polygonum cuspidatum Extract (Pc-Ex) Containing Emodin Suppresses Lung Cancer-Induced Cachexia by Suppressing TCF4/TWIST1 Complex-Induced PTHrP Expression" Nutrients 14, no. 7: 1508. https://doi.org/10.3390/nu14071508