

Vitamin K2 Modulates Mitochondrial Dysfunction Induced by 6-Hydroxydopamine in SH-SY5Y Cells via Mitochondrial Quality-Control Loop


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drug Treatment
2.3. Cell Viability Assay
2.4. Reactive Oxygen Species (ROS) Assessment
2.5. Measurement of Apoptosis
2.6. Detection of Mitochondrial Membrane Potential
2.7. Real-Time Fluorescent Quantitative PCR Analysis
2.8. Western Blotting
2.9. Statistical Analyses
3. Results
3.1. Vitamin K2 Protects SH-SY5Y Cells from 6-OHDA-Induced Reduction in Cell Viability
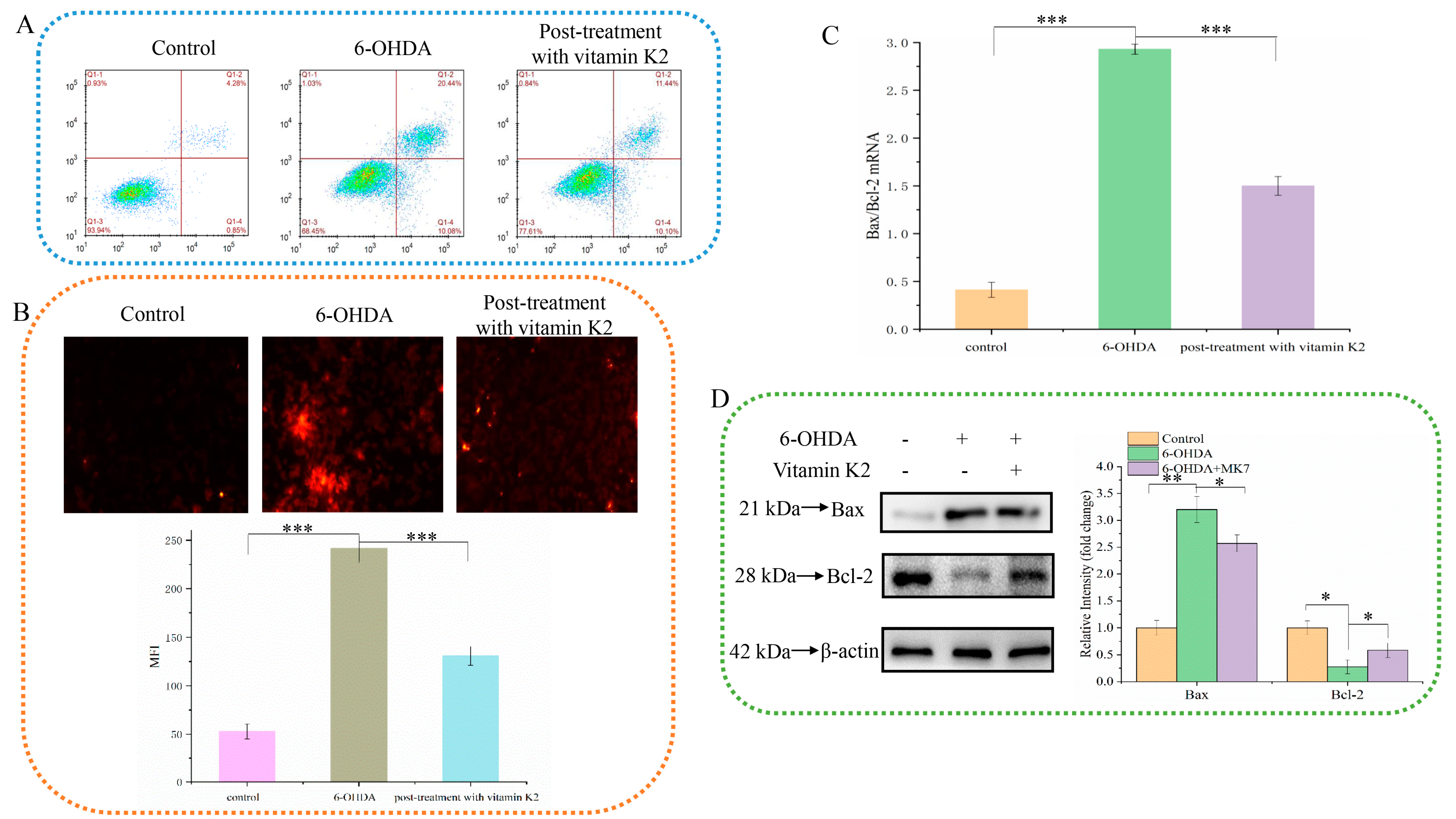
3.2. Vitamin K2 Has a Protective Effect on 6-OHDA-Mediated Apoptosis of SH-SY5Y Cells
3.3. Vitamin K2 Relieves Oxidative Stress Caused by 6-OHDA
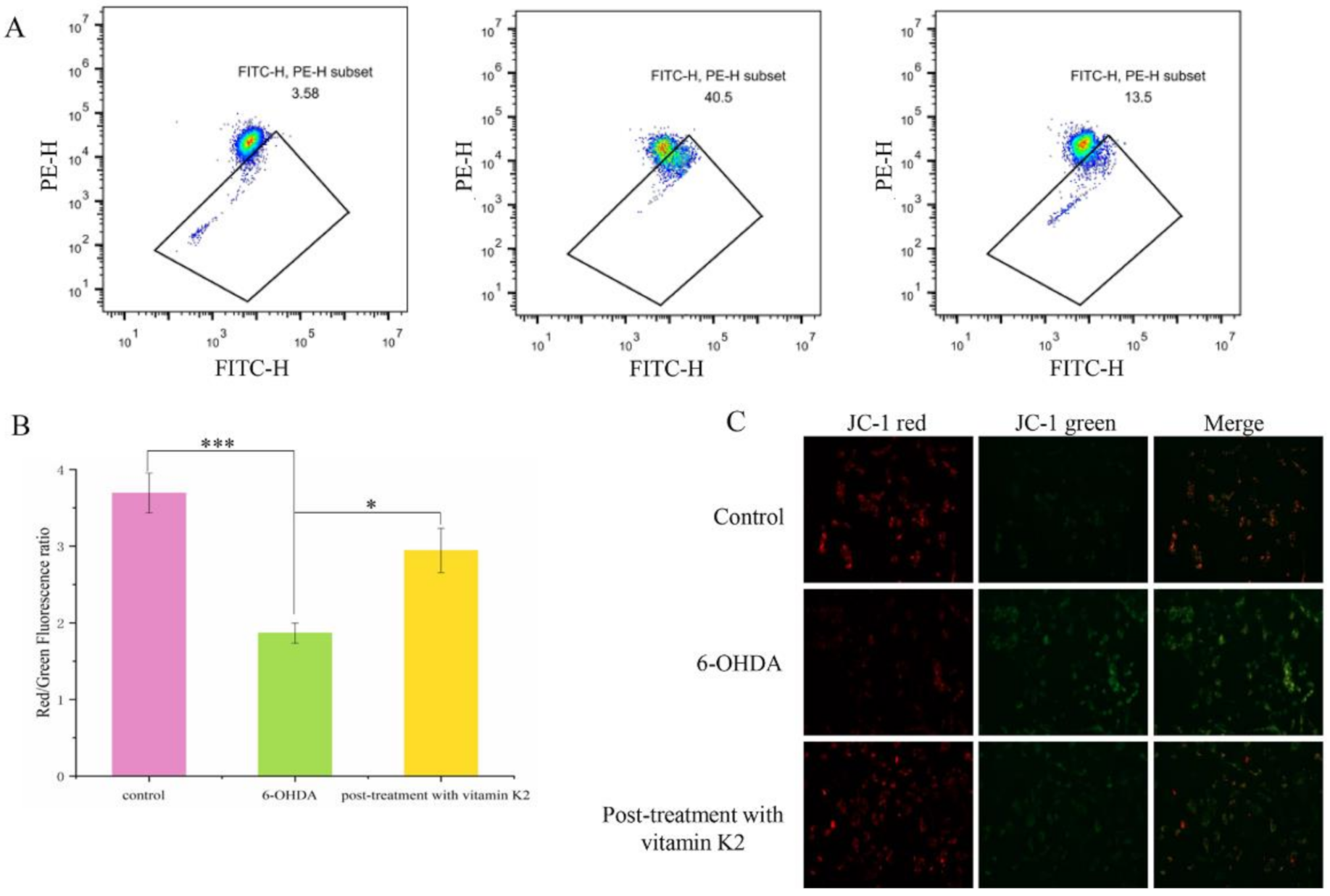
3.4. Vitamin K2 Inhibits Mitochondrial Depolarization Induced by 6-OHDA
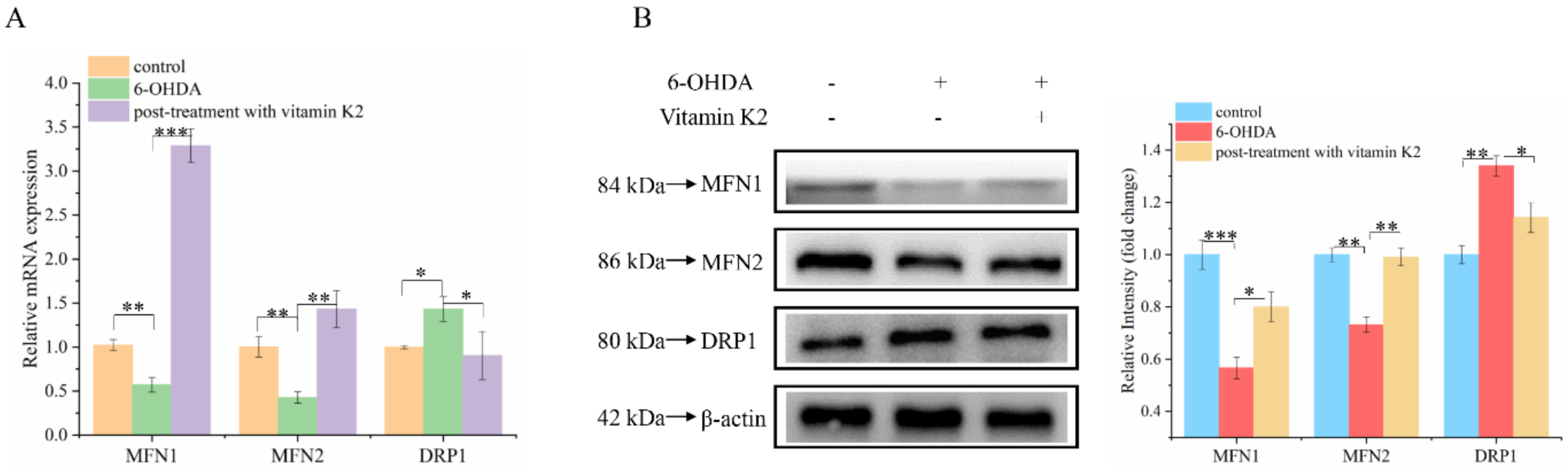
3.5. Effects of Vitamin K2 on Mitochondrial Fusion and Division in 6-OHDA-Mediated Injury Cells
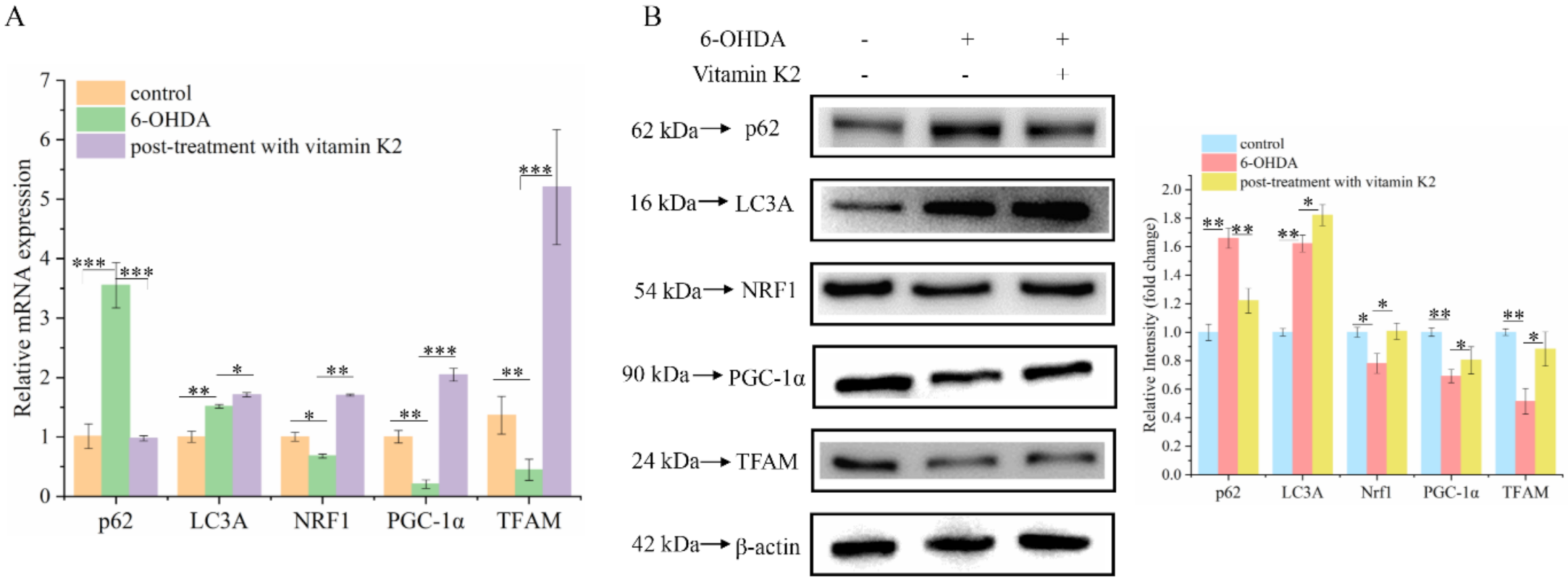
3.6. Effects of Vitamin K2 on Mitophagy and Mitochondrial Biogenesis in 6-OHDA-Mediated Injury Cells
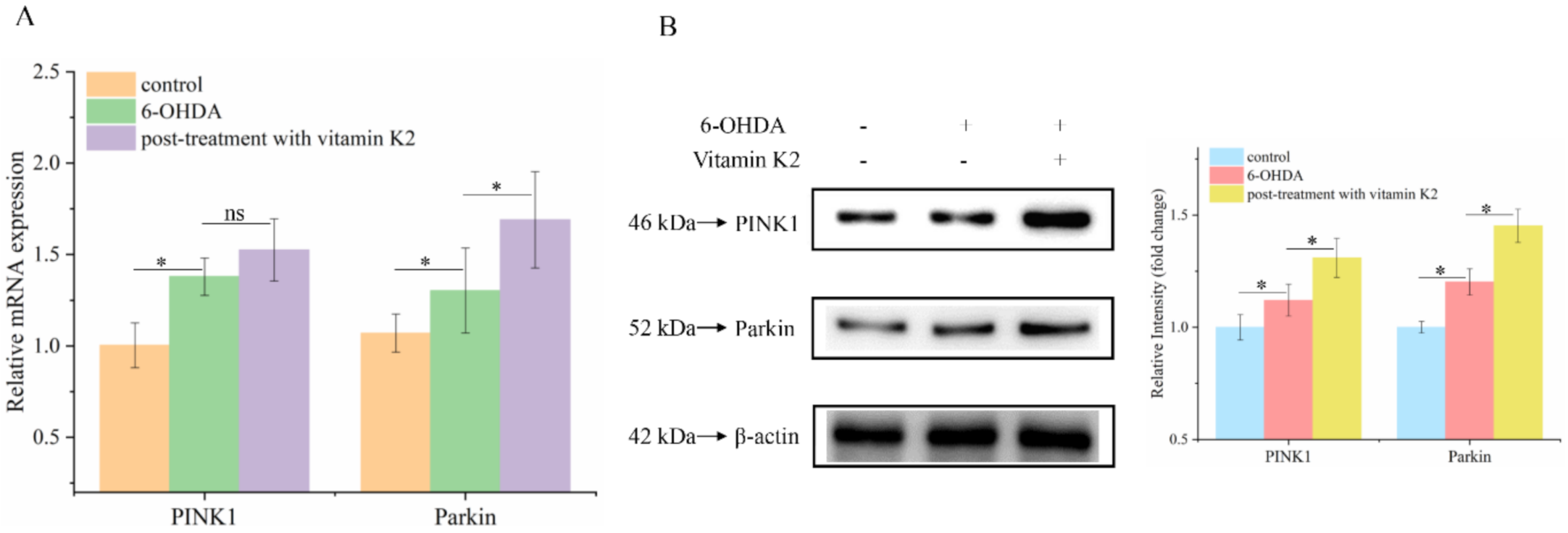
3.7. Vitamin K2 Regulates Mitochondrial Quality-Control System by Activating Pink1/Parkin Signaling Pathway
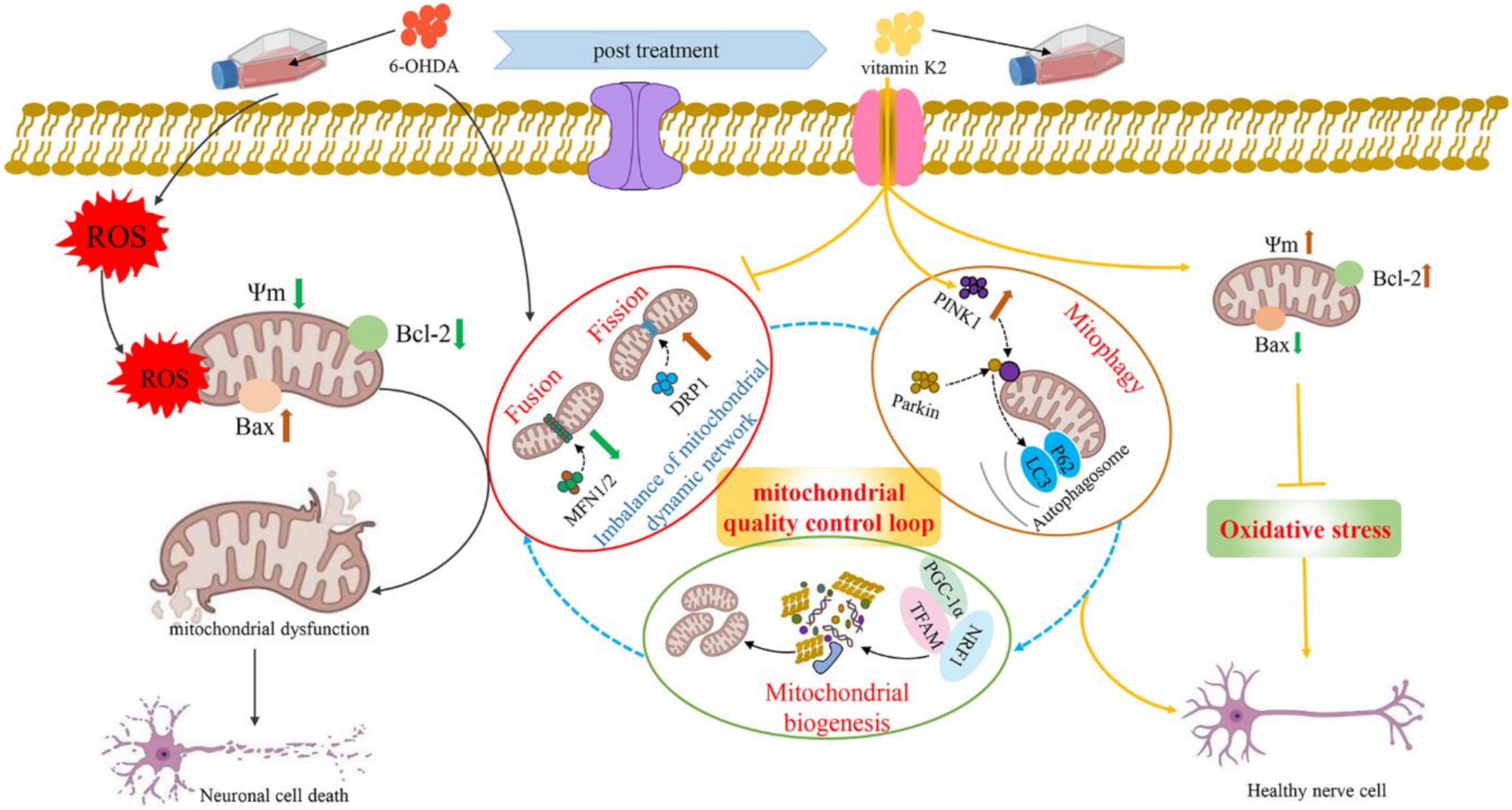
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corrada, M.; Hayden, K.; Paganini-Hill, A.; Bullain, S.; DeMoss, J.; Aguirre, C.; Brookmeyer, R.; Kawas, C. Age of onset of hypertension and risk of dementia in the oldest-old: The 90+ Study. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2017, 13, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.; Pankratz, N.; Lill, C.; Do, C.; Hernandez, D.; Saad, M.; DeStefano, A.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Liang, Y.; Schools, S.; Dawson, V.; Dawson, T.; Savitt, J. Development and characterization of a new Parkinson’s disease model resulting from impaired autophagy. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 16503–16509. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.; Hoek, S.; Fon, E.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem.Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Cebrián, C.; Loike, J.; Sulzer, D. Neuroinflammation in Parkinson’s disease animal models: A cell stress response or a step in neurodegeneration? Curr. Top. Behav. Neurosci. 2015, 22, 237–270. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Trempe, J.; Fon, E. Structure and Function of Parkin, PINK1, and DJ-1, the Three Musketeers of Neuroprotection. Front. Neurol. 2013, 4, 38. [Google Scholar] [CrossRef]
- Lee, C.; Liu, C.; Hsieh, R.; Wei, Y. Oxidative stress-induced depolymerization of microtubules and alteration of mitochondrial mass in human cells. Ann. N. Y. Acad. Sci. 2005, 1042, 246–254. [Google Scholar] [CrossRef]
- Graziewicz, M.; Day, B.; Copeland, W. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res. 2002, 30, 2817–2824. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Mao, K.; Klionsky, D. Mitochondrial fission facilitates mitophagy in Saccharomyces cerevisiae. Autophagy 2013, 9, 1900–1901. [Google Scholar] [CrossRef] [PubMed]
- Anzell, A.; Maizy, R.; Przyklenk, K.; Sanderson, T. Mitochondrial Quality Control and Disease: Insights into Ischemia-Reperfusion Injury. Mol. Neurobiol. 2018, 55, 2547–2564. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.; Grenier, K.; Aguileta, M.; Muise, S.; Farazifard, R.; Haque, M.; McBride, H.; Park, D.; Fon, E. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Hirota, Y.; Nakagawa, K.; Sawada, N.; Okuda, N.; Suhara, Y.; Uchino, Y.; Kimoto, T.; Funahashi, N.; Kamao, M.; Tsugawa, N.; et al. Functional characterization of the vitamin K2 biosynthetic enzyme UBIAD1. PLoS ONE 2015, 10, e0125737. [Google Scholar] [CrossRef]
- Shearer, M.; Newman, P. Metabolism and cell biology of vitamin K. Thromb. Haemost. 2008, 100, 530–547. [Google Scholar] [PubMed]
- Simes, D.; Viegas, C.; Araújo, N.; Marreiros, C. Vitamin K as a Diet Supplement with Impact in Human Health: Current Evidence in Age-Related Diseases. Nutrients 2020, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Pan, J.; Chen, Y.; Li, Y.; Wu, J.; Wang, X. Menaquinone-7 protects astrocytes by regulating mitochondrial function and inflammatory response under hypoxic conditions. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 10181–10193. [Google Scholar] [CrossRef]
- Vos, M.; Esposito, G.; Edirisinghe, J.; Vilain, S.; Haddad, D.; Slabbaert, J.; Van Meensel, S.; Schaap, O.; De Strooper, B.; Meganathan, R.; et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science 2012, 336, 1306–1310. [Google Scholar] [CrossRef]
- Yu, Y.; Li, Y.; Gao, F.; Hu, Q.; Zhang, Y.; Chen, D.; Wang, G. Vitamin K2 suppresses rotenone-induced microglial activation in vitro. Acta Pharmacol. Sin. 2016, 37, 1178–1189. [Google Scholar] [CrossRef]
- Ko, L.; Liu, W.; Georgieff, I.; Yen, S. Modulated induction of tau proteins in cultured human neuroblastoma cells. Brain Res. 1996, 707, 256–265. [Google Scholar] [CrossRef]
- Martinez, T.; Greenamyre, J. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.; Wang, X.; Hu, W.; et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.; Youle, R. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Michel, P.; Hirsch, E.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Hadipour, E.; Tayarani-Najaran, Z.; Fereidoni, M. Vitamin K2 protects PC12 cells against Aβ and HO-induced apoptosis via p38 MAP kinase pathway. Nutr. Neurosci. 2020, 23, 343–352. [Google Scholar] [CrossRef]
- Huang, S.; Fang, S.; Chen, Y. Molecular Mechanism of Vitamin K2 Protection against Amyloid-β-Induced Cytotoxicity. Biomolecules 2021, 11, 423. [Google Scholar] [CrossRef]
- Shandilya, S.; Kesari, K.; Ruokolainen, J. Vitamin K2 Modulates Organelle Damage and Tauopathy Induced by Streptozotocin and Menadione in SH-SY5Y Cells. Antioxidants 2021, 10, 983. [Google Scholar] [CrossRef]
- Shadel, G.; Horvath, T. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef]
- Gu, X.; Manautou, J. Molecular mechanisms underlying chemical liver injury. Expert Rev. Mol. Med. 2012, 14, e4. [Google Scholar] [CrossRef]
- Ferger, B.; Themann, C.; Rose, S.; Halliwell, B.; Jenner, P. 6-hydroxydopamine increases the hydroxylation and nitration of phenylalanine in vivo: Implication of peroxynitrite formation. J. Neurochem. 2001, 78, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Shirakabe, A.; Brady, C.; Zablocki, D.; Ohishi, M.; Sadoshima, J. Molecular mechanisms mediating mitochondrial dynamics and mitophagy and their functional roles in the cardiovascular system. J. Mol. Cell. Cardiol. 2015, 78, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. CMLS 2016, 73, 775–795. [Google Scholar] [CrossRef]
- Sun, Y.; Vashisht, A.; Tchieu, J.; Wohlschlegel, J.; Dreier, L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J. Biol. Chem. 2012, 287, 40652–40660. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schlehe, J.; LaVoie, M.; Schwarz, T. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Gomes, A.; Price, N.; Ling, A.; Moslehi, J.; Montgomery, M.; Rajman, L.; White, J.; Teodoro, J.; Wrann, C.; Hubbard, B.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- Ham, P.; Raju, R. Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog. Neurobiol. 2017, 157, 92–116. [Google Scholar] [CrossRef]
- Mollica, G.; Senesi, P.; Codella, R.; Vacante, F.; Montesano, A.; Luzi, L.; Terruzzi, I. L-carnitine supplementation attenuates NAFLD progression and cardiac dysfunction in a mouse model fed with methionine and choline-deficient diet. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2020, 52, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, H.; Zheng, Z.; Wang, H.; Wang, L.; Zhao, G.; Wang, P. Vitamin K2 Modulates Mitochondrial Dysfunction Induced by 6-Hydroxydopamine in SH-SY5Y Cells via Mitochondrial Quality-Control Loop. Nutrients 2022, 14, 1504. https://doi.org/10.3390/nu14071504
Tang H, Zheng Z, Wang H, Wang L, Zhao G, Wang P. Vitamin K2 Modulates Mitochondrial Dysfunction Induced by 6-Hydroxydopamine in SH-SY5Y Cells via Mitochondrial Quality-Control Loop. Nutrients. 2022; 14(7):1504. https://doi.org/10.3390/nu14071504
Chicago/Turabian StyleTang, Hengfang, Zhiming Zheng, Han Wang, Li Wang, Genhai Zhao, and Peng Wang. 2022. "Vitamin K2 Modulates Mitochondrial Dysfunction Induced by 6-Hydroxydopamine in SH-SY5Y Cells via Mitochondrial Quality-Control Loop" Nutrients 14, no. 7: 1504. https://doi.org/10.3390/nu14071504