
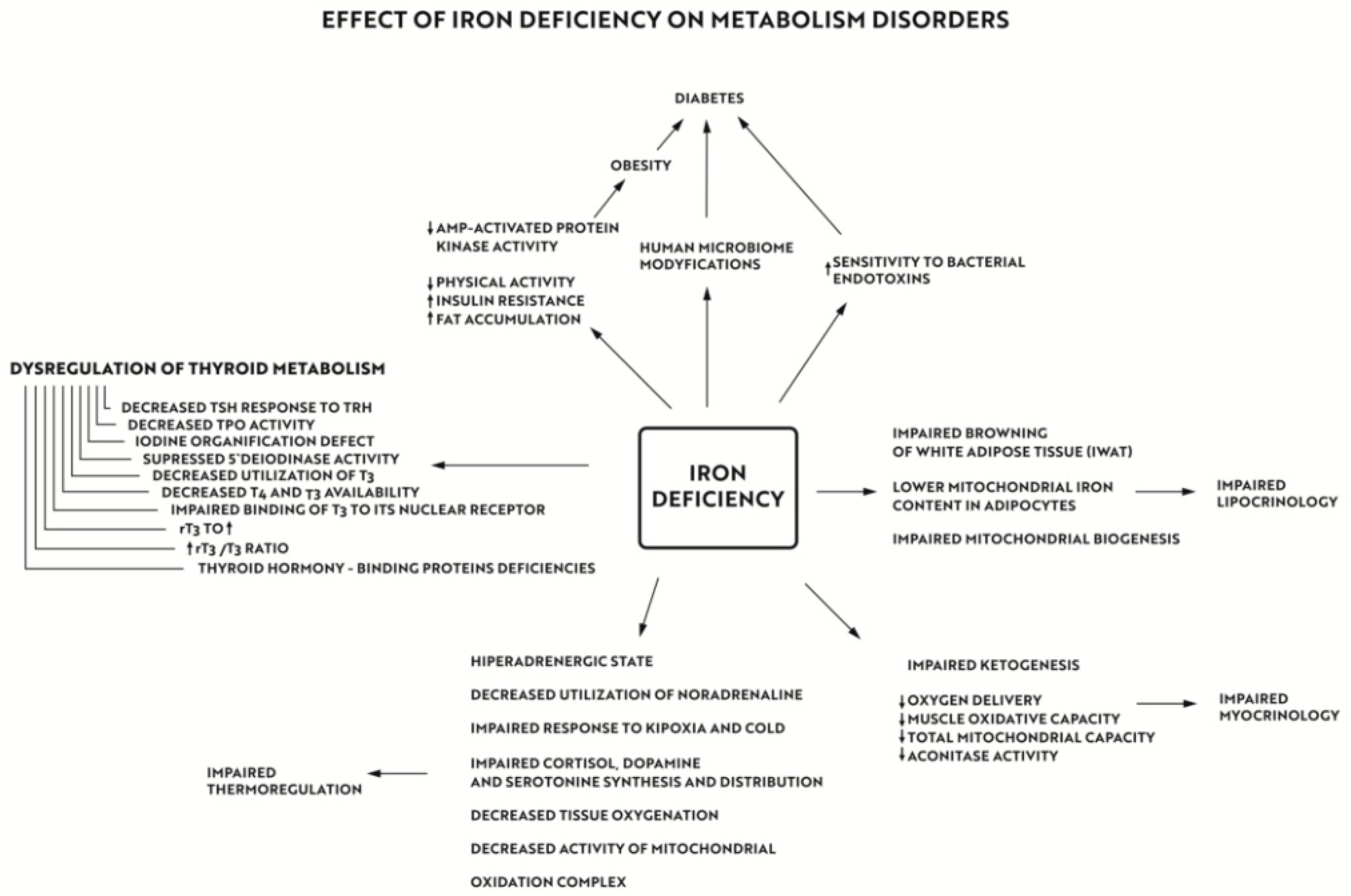
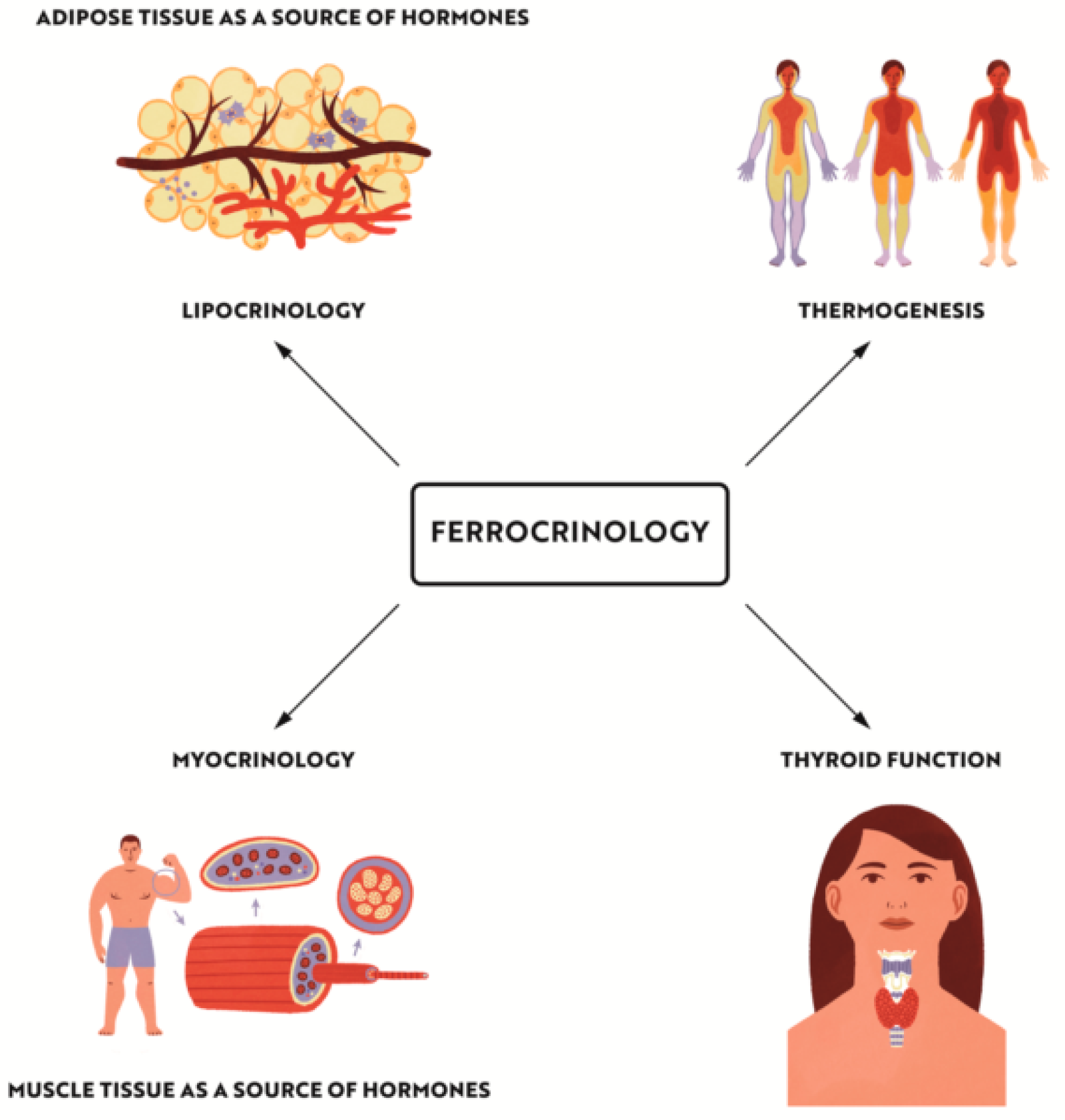
“Ferrocrinology”—Iron Is an Important Factor Involved in Gluco- and Lipocrinology
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Introduction to Ferrocrinology—Effects of Iron Deficiency on Thermoregulation and Stress Response
2.1. ID Impairs Hormonal Response to Hypoxia
2.2. Iron Deficiency Impairs the Utilization of Norepinephrine and Favours the Hyperadrenergic State
2.3. ID Impairs TSH Secretion
2.4. ID Results in Impaired Binding of Triiodothyronine to Its Nuclear Receptor
2.5. ID Can Lead to Overgrowth and Dysfunction of IBAT
2.6. How Iron Deficiency can Lead to Increased Senstitivty to Stress
3. Effect of Iron Deficiency on Autoimmune Thyroid Disease (Aitd) Development
3.1. AITD—The Most Prevalent Endocrinopathy
3.2. A Link between Thyroid Disorders and Development of Obesity and Diabetes—Mutual Interactions
3.3. How ID Can Contribute to the Increased Risk of AITD?—Possible Pathogenetic Mechanisms
4. The Impact of Iron Deficiency on Obesity and Diabetes
4.1. How Iron Deficiency can Contribute to the Development of Obesity—General Interactions
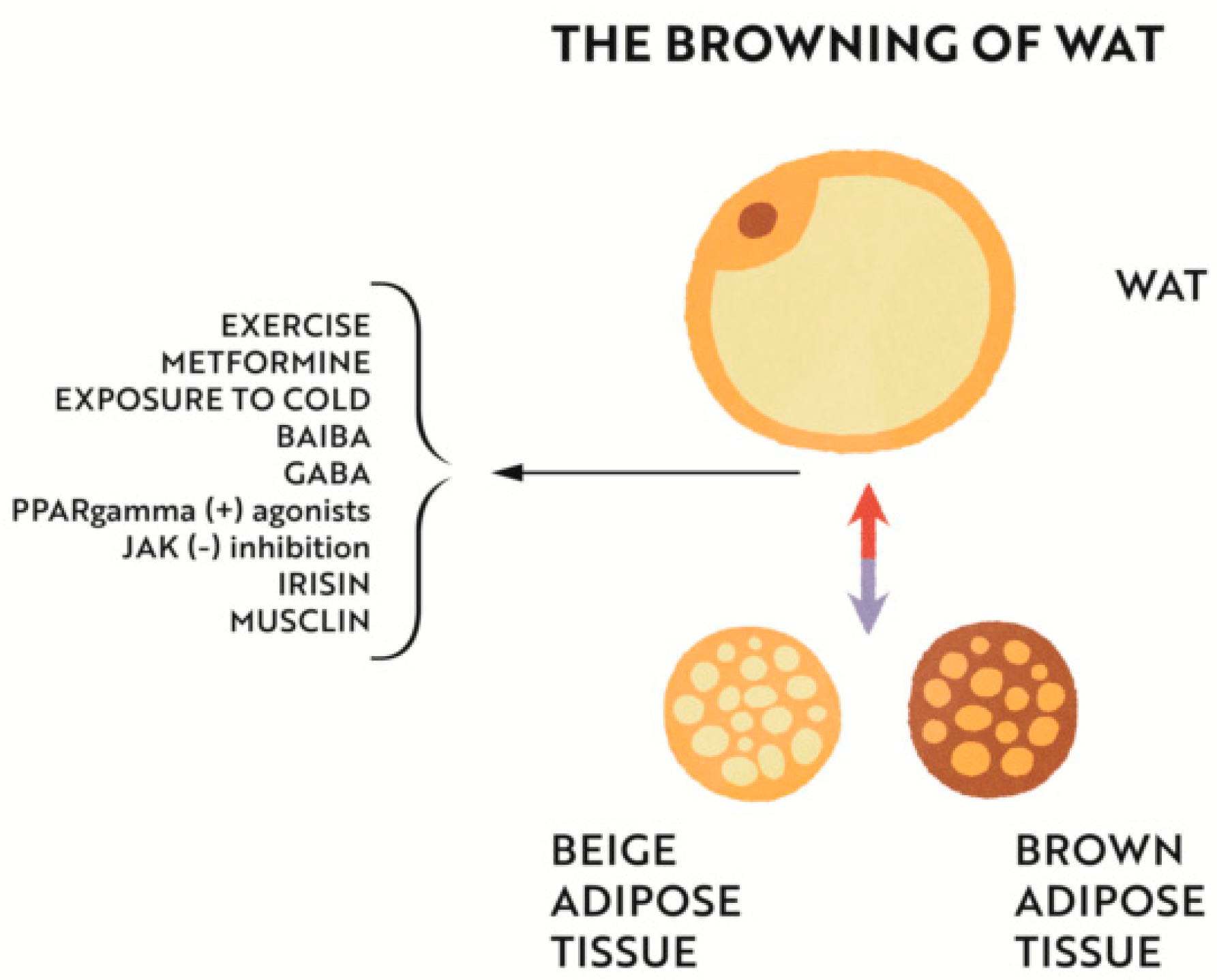
4.2. Iron Effect on Adipose and Muscle Tissue Metabolism
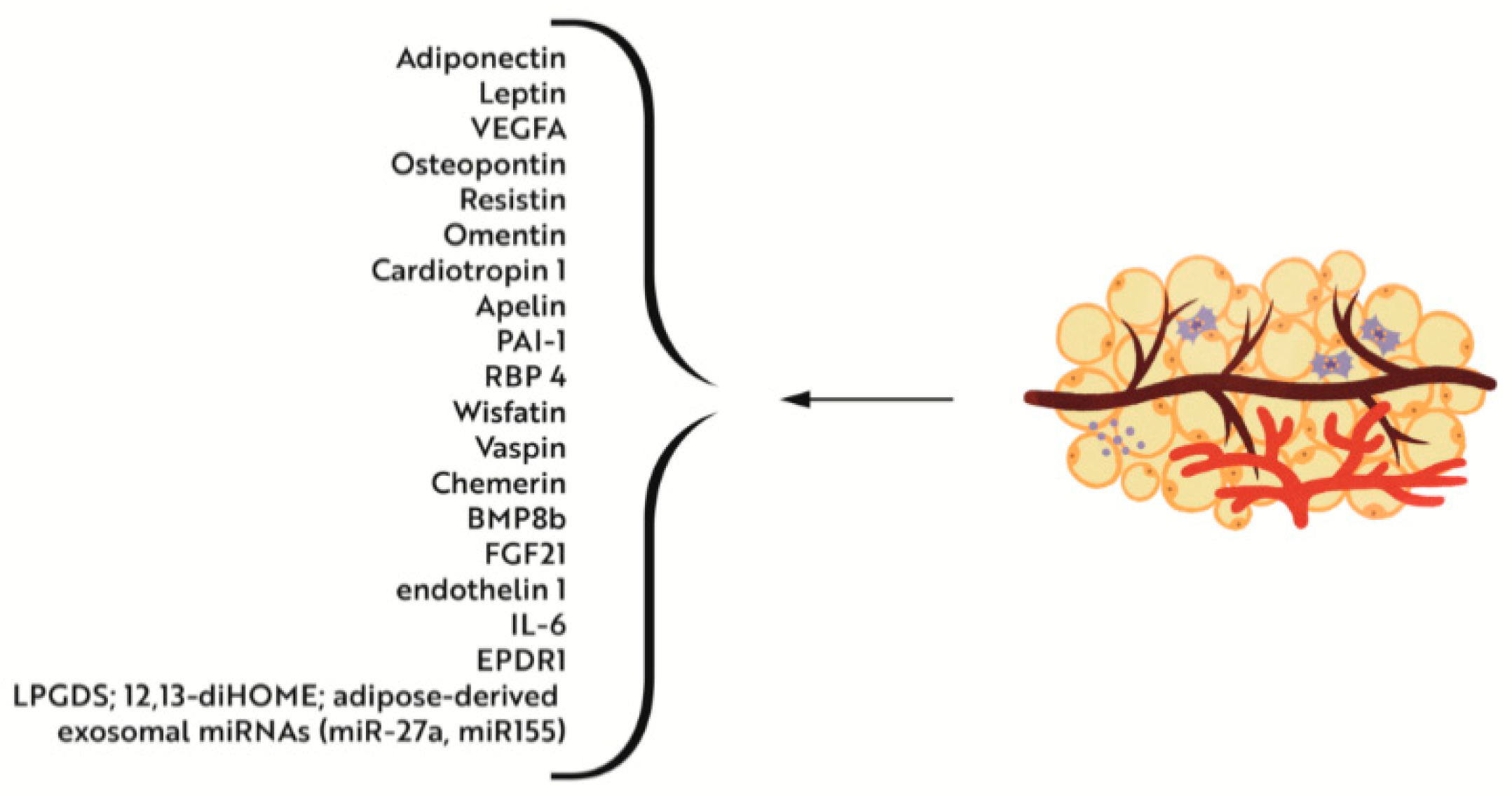
4.2.1. Iron Deficiency versus Adipose Tissue Metabolism and Interactions with Muscle Tissue
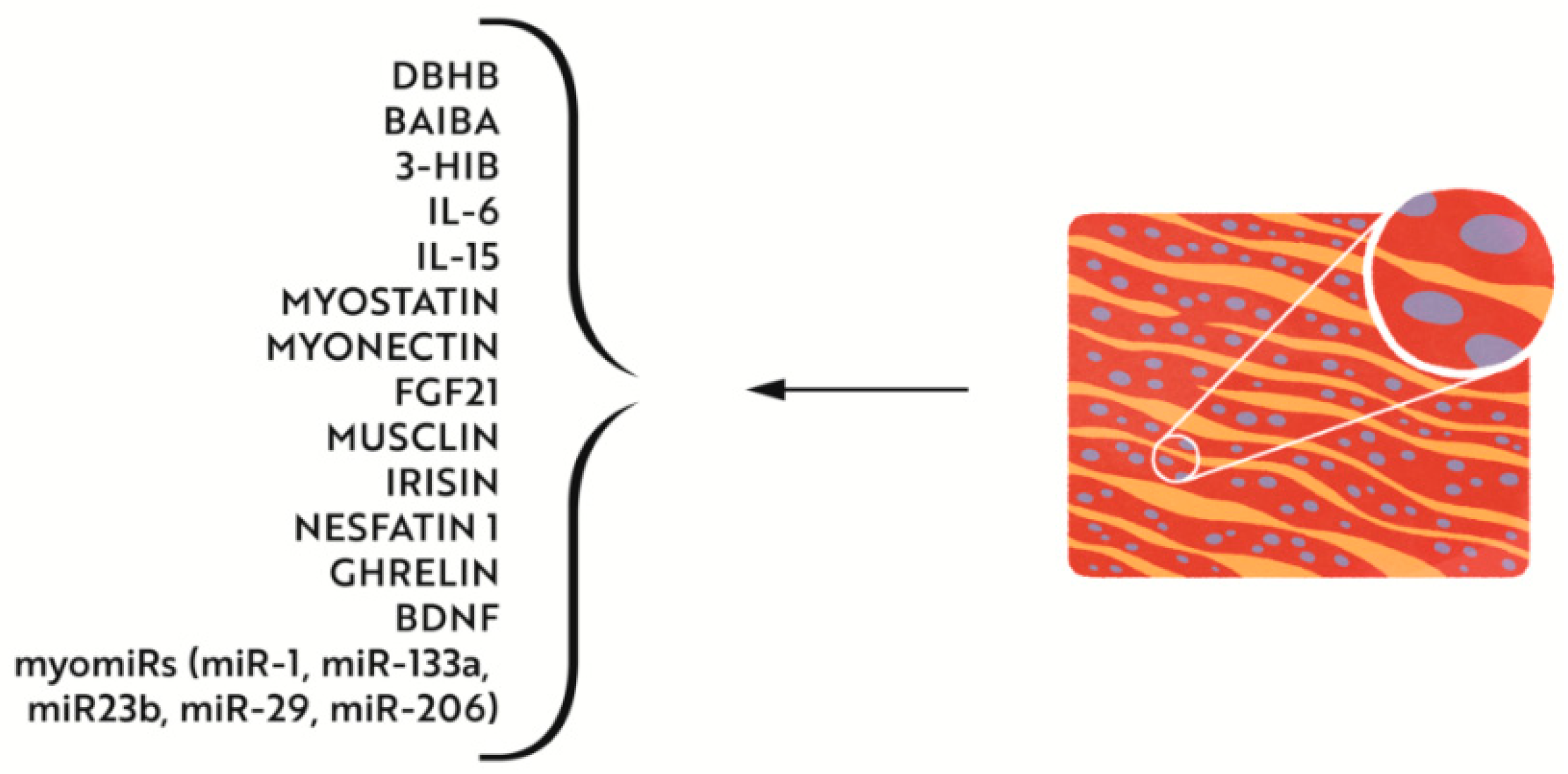
4.2.2. Effects of Iron Deficiency on Muscle Metabolism
4.3. Effects of Iron Deficiency on the Development of Type 2 Diabetes Mellitus
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ID | iron deficiency |
| IDA | iron deficiency anaemia |
| AITD | autoimmune thyroid disease |
References
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Wang, R.; Shan, Z.; Dong, Y.; Zheng, H.; Jesse, F.F.; Rao, E.; Takahashi, E.; Li, W.; Teng, W.; et al. Perinatal Iron Deficiency-Induced Hypothyroxinemia Impairs Early Brain Development Regardless of Normal Iron Levels in the Neonatal Brain. Thyroid 2016, 26, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Cacoub, P.; Macdougall, I.C.; Peyrin-Biroulet, L. Iron deficiency anaemia. Lancet 2015, 387, 907–916. [Google Scholar] [CrossRef]
- Kalra, S.; Priya, G.; Gupta, Y. Glucocrinology. J. Pak. Med. Assoc. 2018, 68, 963–965. [Google Scholar] [PubMed]
- Kalra, S.; Priya, G. Lipocrinology—The relationship between lipids and endocrine function. Drugs Context 2018, 7, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okla, M.; Kim, J.; Koehler, K.; Chung, S. Dietary Factors Promoting Brown and Beige Fat Development and Thermogenesis. Adv. Nutr. 2017, 8, 473–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, H.; Bonora, M.; M’Barek, S.B.; Sinclair, J.D. Effects of hypoxia and cold acclimation on thermoregulation in the rat. J. Appl. Physiol. 1991, 71, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Surks, M.I. Effect of thyrotropin on thyroidal iodine metabolism during hypoxia. Am. J. Physiol. 1969, 216, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Gordon, C.J.; Fogelson, L. Comparative effects of hypoxia on behavioral thermoregulation in rats, hamsters, and mice. Am. J. Physiol. 1991, 260, R120–R125. [Google Scholar] [CrossRef]
- Galton, V.A. Some Effects of Altitude on Thyroid Function. Endocrinology 1972, 91, 1393–1403. [Google Scholar] [CrossRef]
- Moshang, T.; Chance, K.H.; Kaplan, M.M.; Utiger, R.D.; Takahashi, O. Effects of hypoxia on thyroid function tests. J. Pediatr. 1980, 97, 602–604. [Google Scholar] [CrossRef]
- Mayfield, S.R.; Shaul, P.W.; Oh, W.; Stonestreet, B.S. Anemia Blunts the Thermogenic Response to Environmental Cold Stress in Newborn Piglets. Pediatr. Res. 1987, 21, 482–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillman, E.; Galé, C.; Green, W.; Johnson, D.G.; Mackler, B.; Finch, C. Hypothermia in iron deficiency due to altered triiodothyronine metabolism. Am. J. Physiol. 1980, 239, R377–R381. [Google Scholar] [CrossRef] [PubMed]
- Brigham, D.; Beard, J.; Tobin, B. Iron and thermoregulation: A review. Crit. Rev. Food Sci. Nutr. 1996, 36, 747–763. [Google Scholar] [CrossRef]
- Sato, T.; Miyamori, C.; Kajiwara, S.; Murata, A. The role of thyrotropin-releasing hormone (TRH) and histidyl-proline diketopiperazine (HPD) in the maturation of thermogenesis in young rats. Pediatr. Res. 1985, 19, 614. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.E. Pituitary—Thyroid relationships in hypothyroidism. Baillière’s Clin. Endocrinol. Metab. 1988, 2, 541–565. [Google Scholar] [CrossRef]
- Malbon, C.C.; Graziano, M.P.; Johnson, G.L. Fat cell beta-adrenergic receptor in the hypothyroid rat. Impaired interaction with the stimulatory regulatory component of adenylate cyclase. J. Biol. Chem. 1984, 259, 3254–3260. [Google Scholar] [CrossRef]
- Goswami, A.; Rosenberg, I.N. Effects of thyroid status on membrane-bound low Km cyclic nucleotide phosphodiesterase activities in rat adipocytes. J. Biol. Chem. 1985, 260, 82–85. [Google Scholar] [CrossRef]
- Beard, J.; Tobin, B.; Green, W. Evidence for Thyroid Hormone Deficiency in Iron-Deficient Anemic Rats. J. Nutr. 1989, 119, 772–778. [Google Scholar] [CrossRef]
- Beard, J. Feed Efficiency and Norepinephrine Turnover in Iron Deficiency. Exp. Biol. Med. 1987, 184, 337–344. [Google Scholar] [CrossRef]
- Raasmaja, A.; Larsen, P.R. α1- and β-Adrenergic Agents Cause Synergistic Stimulation of the Iodothyronine Deiodinase in Rat Brown Adipocytes*. Endocrinology 1989, 125, 2502–2509. [Google Scholar] [CrossRef] [PubMed]
- Dillmann, E.; Johnson, D.G.; Martín, J.; Mackler, B.; Finch, C. Catecholamine elevation in iron deficiency. Am. J. Physiol. 1979, 237, R297–R300. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.A.; Beard, J.L.; Druckenmiller, M. Increased Insulin Sensitivity in Iron-Deficient Rats. J. Nutr. 1988, 118, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.L.; Tobin, B.W.; Smith, S.M. Effects of Iron Repletion and Correction of Anemia on Norepinephrine Turnover and Thyroid Metabolism in Iron Deficiency. Exp. Biol. Med. 1990, 193, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Baum, D.; Porte, D.; Ensinck, J. Hyperglucagonemia and alpha-adrenergic receptor in acute hypoxia. Am. J. Physiol. 1979, 237, E404. [Google Scholar] [CrossRef]
- Cherrington, A.D.; Fuchs, H.; Stevenson, R.W.; Williams, P.E.; Alberti, K.G.; Steiner, K.E. Effect of epinephrine on glycogenolysis and gluconeogenesis in conscious overnight-fasted dogs. Am. J. Physiol. 1984, 247, E137–E144. [Google Scholar] [CrossRef]
- Natalicchio, A.; Marrano, N.; Biondi, G.; Spagnuolo, R.; Labarbuta, R.; Porreca, I.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; Perrini, S.; et al. The Myokine Irisin Is Released in Response to Saturated Fatty Acids and Promotes Pancreatic β-Cell Survival and Insulin Secretion. Diabetes 2017, 66, 2849–2856. [Google Scholar] [CrossRef] [Green Version]
- Voorhess, M.L.; Stuart, M.J.; Stockman, J.A.; Oski, F.A. Iron deficiency anemia and increased urinary norepinephrine excretion. J. Pediatr. 1975, 86, 542–547. [Google Scholar] [CrossRef]
- Wagner, A.; Fortier, N.; Giroux, A.; Lukes, J.; Snyder, L.M. Catecholamines in adult iron deficiency patients. Experientia 1979, 35, 681–682. [Google Scholar] [CrossRef]
- Beard, J.; Green, W.; Miller, L.; Finch, C. Effect of iron-deficiency anemia on hormone levels and thermoregulation during cold exposure. Am. J. Physiol. 1984, 247, R114–R119. [Google Scholar] [CrossRef]
- Tang, F.; Wong, T.M.; Loh, T.T. Effects of Cold Exposure or TRH on the Serum TSH Levels in the Iron-Deficient Rat. Horm. Metab. Res. 1988, 20, 616–619. [Google Scholar] [CrossRef]
- Beard, J.L.; Chen, Q.; Connor, J.; Jones, B.C. Altered monamine metabolism in caudate-putamen of iron-deficient rats. Pharmacol. Biochem. Behav. 1994, 48, 621–624. [Google Scholar] [CrossRef]
- Youdim, M.B.; Ben-Shachar, D.; Yehuda, S. Putative biological mechanisms of the effect of iron deficiency on brain biochemistry and behavior. Am. J. Clin. Nutr. 1989, 50, 607–617, Erratum in Am. J. Clin. Nutr. 1990, 5, 319. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Yehuda, S. Iron deficiency induces reversal of dopamine dependent circadian cycles: Differential response to d-amphetamine and TRH. Peptides 1985, 6, 851–855. [Google Scholar] [CrossRef]
- Beard, J.L.; Brigham, D.E.; Kelley, S.K.; Green, M.H. Plasma thyroid hormone kinetics are altered in iron-deficient rats. J. Nutr. 1998, 128, 1401–1408. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, G.; A Bittle, P.; Sanders, H.; Bercu, B.B. Hypothalamo-hypophyseal thyroid and gonadal function before and after erythropoietin therapy in dialysis patients. J. Clin. Endocrinol. Metab. 1992, 74, 517–524. [Google Scholar] [CrossRef]
- Scammell, J.G.; Shiverick, K.T.; Fregly, M.J. In vitro hepatic deiodination of L-thyroxine to 3,5,3’-triiodothyronine in cold-acclimated rats. J. Appl. Physiol. 1980, 49, 386–389. [Google Scholar] [CrossRef]
- Wasserman, D.H.; Lavina, H.; Lickley, A.; Vranić, M. Effect of hematocrit reduction on hormonal and metabolic responses to exercise. J. Appl. Physiol. 1985, 58, 1257–1262. [Google Scholar] [CrossRef]
- Silva, J.E.; Larsen, P.R. Potential of brown adipose tissue type II thyroxine 5’-deiodinase as a local and systemic source of triiodothyronine in rats. J. Clin. Investig. 1985, 76, 2296–2305. [Google Scholar] [CrossRef]
- Bastian, T.W.; Prohaska, J.R.; Georgieff, M.K.; Anderson, G.W. Perinatal Iron and Copper Deficiencies Alter Neonatal Rat Circulating and Brain Thyroid Hormone Concentrations. Endocrinology 2010, 151, 4055–4065. [Google Scholar] [CrossRef]
- Martínez-Torres, C.; Cubeddu, L.; Dillmann, E.; Brengelmann, G.L.; Leets, I.; Layrisse, M.; Johnson, D.G.; Finch, C. Effect of exposure to low temperature on normal and iron-deficient subjects. Am. J. Physiol. 1984, 246, R380–R383. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.L.; Borel, M.J.; Derr, J. Impaired thermoregulation and thyroid function in iron-deficiency anemia. Am. J. Clin. Nutr. 1990, 52, 813–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iddah, M.A.; Macharia, B.N. Autoimmune Thyroid Disorders. ISRN Endocrinol. 2013, 2013, 509764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallman, M.F. Stress-induced obesity and the emotional nervous system. Trends Endocrinol. Metab. 2010, 21, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Pouwer, F.; Kupper, N.; Adriaanse, M.C. Does emotional stress cause type 2 diabetes mellitus? A review from the European Depression in Diabetes (EDID) Research Consortium. Discov. Med. 2010, 9, 112–118. [Google Scholar]
- Lukaski, H.C.; Hall, C.B.; Nielsen, F.H. Thermogenesis and thermoregulatory function of iron-deficient women without anemia. Aviat. Space Environ. Med. 1990, 61, 913–920. [Google Scholar] [PubMed]
- Coleman, R.; Tanne, Z.; Nahir, M.; Shomrat, D.; Miller-Lotan, R.; Youdim, M. Ultrastructural Changes in Mitochondria of the Adrenal Cortex of Iron-Deficient Rats. Cells Tissues Organs 1995, 152, 33–40. [Google Scholar] [CrossRef]
- Lieu, P.T.; Heiskala, M.; Peterson, P.A.; Yang, Y. The roles of iron in health and disease. Mol. Asp. Med. 2001, 22, 1–87. [Google Scholar] [CrossRef]
- de Castro, J.P.W.; Fonseca, T.L.; Ueta, C.B.; McAninch, E.A.; Abdalla, S.; Wittmann, G.; Lechan, R.M.; Gereben, B.; Bianco, A.C. Differences in hypothalamic type 2 deiodinase ubiquitination explain localized sensitivity to thyroxine. J. Clin. Investig. 2015, 125, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Youdim, M.B.; Green, A.R.; Bloomfield, M.R.; Mitchell, B.D.; Heal, D.J.; Grahame-Smith, D.G. The effects of iron deficiency on brain biogenic monoamine biochemistry and function in rats. Neuropharmacology 1980, 19, 259–267. [Google Scholar] [CrossRef]
- Rots, N.Y.; De Jong, J.; Workel, J.O.; Levine, S.; Cools, A.R.; De Kloet, E.R. Neonatal Maternally Deprived Rats have as Adults Elevated Basal Pituitary-Adrenal Activity and Enhanced Susceptibility to Apomorphine. J. Neuroendocr. 1996, 8, 501–506. [Google Scholar] [CrossRef]
- Mahajan, S.D.; Singh, S.; Shah, P.; Gupta, N.; Kochupillai, N. Effect of Maternal Malnutrition and Anemia on the Endocrine Regulation of Fetal Growth. Endocr. Res. 2004, 30, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, F.; Fallahi, P.; Elia, G.; Gonnella, D.; Paparo, S.R.; Giusti, C.; Churilov, L.P.; Ferrari, S.M.; Antonelli, A. Hashimotos’ thyroiditis: Epidemiology, pathogenesis, clinic and therapy. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101367. [Google Scholar] [CrossRef] [PubMed]
- McLeod, D.S.; Cooper, D.S. The incidence and prevalence of thyroid autoimmunity. Endocrine 2012, 42, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferrari, S.M.; Corrado, A.; Di Domenicantonio, A.; Fallahi, P. Autoimmune thyroid disorders. Autoimmun. Rev. 2015, 14, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.M.; Fallahi, P.; Antonelli, A.; Benvenga, S. Environmental Issues in Thyroid Diseases. Front. Endocrinol. 2017, 8, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, M.B. The Influence of Iron Status on Iodine Utilization and Thyroid Function. Annu. Rev. Nutr. 2006, 26, 367–389. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Bi, Y.; Cui, B.; Hong, J.; Wang, W.; Ning, G. The New Perspectives on Genetic Studies of Type 2 Diabetes and Thyroid Diseases. Curr. Genom. 2013, 14, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Hage, M.; Zantout, M.S.; Azar, S.T. Thyroid Disorders and Diabetes Mellitus. J. Thyroid Res. 2011, 2011, 439463. [Google Scholar] [CrossRef] [Green Version]
- Al-Geffari, M.; Ahmad, N.A.; Al-Sharqawi, A.H.; Youssef, A.M.; AlNaqeb, D.; Al-Rubeaan, K. Risk Factors for Thyroid Dysfunction among Type 2 Diabetic Patients in a Highly Diabetes Mellitus Prevalent Society. Int. J. Endocrinol. 2013, 2013, 417920. [Google Scholar] [CrossRef]
- Biondi, B.; Kahaly, G.J.; Robertson, R.P. Thyroid Dysfunction and Diabetes Mellitus: Two Closely Associated Disorders. Endocr. Rev. 2019, 40, 789–824. [Google Scholar] [CrossRef] [PubMed]
- Wartofsky, L. Thyrotoxic storm. In Werner and Ingbar’s The Thyroid; Braverman, L.E., Utiger, R.D., Eds.; Lippincott–Raven: New York, NY, USA, 2000; pp. 679–684. [Google Scholar]
- Dimitriadis, G.; Parry-Billings, M.; Bevan, S.; Leighton, B.; Krause, U.; Piva, T.; Tegos, K.; Challiss, J.; Wegener, G.; Newsholme, E.A. The effects of insulin on transport and metabolism of glucose in skeletal muscle from hyperthyroid and hypothyroid rats. Eur. J. Clin. Investig. 1997, 27, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.O.; Anil, C.; Gursoy, A.; Nar, A.; Inanc, M.; Bozkurt, O.; Tutuncu, N.B. Thyroid volume in patients with glucose metabolism disorders. Arq. Bras. Endocrinol. Metabol. 2014, 58, 824–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díez, J.J.; Iglesias, P. Relationship Between Thyrotropin and Body Mass Index in Euthyroid Subjects. Exp. Clin. Endocrinol. Diabetes 2010, 119, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Gierach, M.; Gierach, J.; Skowrońska, A.; Rutkowska, E.; Spychalska, M.; Pujanek, M.; Junik, R. Hashimoto’s thyroiditis and carbohydrate metabolism disorders in patients hospitalised in the Department of Endocrinology and Diabetology of Ludwik Rydygier Collegium Medicum in Bydgoszcz between 2001 and 2010. Endokrynol. Pol. 2012, 63, 14–17. [Google Scholar]
- Mouradian, M.; Abourizk, N. Diabetes Mellitus and Thyroid Disease. Diabetes Care 1983, 6, 512–520, Erratum in Diabet Med. 2008, 25, 244. [Google Scholar] [CrossRef]
- Chen, H.-S.; Wu, T.-E.J.; Jap, T.-S.; Lu, R.-A.; Wang, M.-L.; Chen, R.-L.; Lin, H.-D. Subclinical hypothyroidism is a risk factor for nephropathy and cardiovascular diseases in Type 2 diabetic patients. Diabet. Med. 2007, 24, 1336–1344. [Google Scholar] [CrossRef]
- Stefanowicz-Rutkowska, M.M.; Matuszewski, W.; Gontarz-Nowak, K.; Bandurska-Stankiewicz, E.M. Is there a relationship between the prevalence of autoimmune thyroid disease and diabetic kidney disease? Open Life Sci. 2021, 16, 611–619. [Google Scholar] [CrossRef]
- Stefanowicz-Rutkowska, M.M.; Matuszewski, W.; Bandurska-Stankiewicz, E.M. Autoimmune Thyroid Disease is Associated with a Lower Prevalence of Diabetic Retinopathy in Patients with Type 1 Diabetic Mellitus. Medicina 2020, 56, 255. [Google Scholar] [CrossRef]
- Prats-Puig, A.; Sitjar, C.; Ribot, R.; Calvo, M.; Clausell-Pomés, N.; Soler-Roca, M.; Soriano-Rodríguez, P.; Osiniri, I.; Ros-Miquel, M.; Bassols, J.; et al. Relative Hypoadiponectinemia, Insulin Resistance, and Increased Visceral Fat in Euthyroid Prepubertal Girls With Low-Normal Serum Free Thyroxine. Obesity 2012, 20, 1455–1461. [Google Scholar] [CrossRef]
- Alevizaki, M.; Saltiki, K.; Voidonikola, P.; Mantzou, E.; Papamichael, C.; Stamatelopoulos, K. Free thyroxine is an independent predictor of subcutaneous fat in euthyroid individuals. Eur. J. Endocrinol. 2009, 161, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Guan, Q.; Liu, Y.; Wang, H.; Xu, W.; Li, X.; Fu, Y.; Gao, L.; Zhao, J.; Wang, X. Role of extrathyroidal TSHR expression in adipocyte differentiation and its association with obesity. Lipids Heal. Dis. 2012, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.J.; Moreno-Navarrete, J.M.; Ribas, V.; Esteve, E.; Rodriguez-Hermosa, J.I.; Ruiz, B.; Peral, B.; Ricart, W.; Zorzano, A.; Fernández-Real, J.M. Subcutaneous Fat Shows Higher Thyroid Hormone Receptor-α1 Gene Expression Than Omental Fat. Obesity 2009, 17, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- Nannipieri, M.; Cecchetti, F.; Anselmino, M.; Camastra, S.; Niccolini, P.; Lamacchia, M.; Rossi, M.; Iervasi, G.; Ferrannini, E. Expression of thyrotropin and thyroid hormone receptors in adipose tissue of patients with morbid obesity and/or type 2 diabetes: Effects of weight loss. Int. J. Obes. 2009, 33, 1001–1006, Erratum in Int. J. Obes. 2010, 34, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Li, C.W.; Hammerstad, S.S.; Stefan, M.; Tomer, Y. Immunogenetics of autoimmune thyroid diseases: A comprehensive review. J. Autoimmun. 2015, 64, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, A.; Tanigawa, K.; Akama, T.; Yoshihara, A.; Ishii, N.; Suzuki, K. Innate Immune Activation and Thyroid Autoimmunity. J. Clin. Endocrinol. Metab. 2011, 96, 3661–3671. [Google Scholar] [CrossRef] [Green Version]
- Dittmar, M.; Kahaly, G.J. Polyglandular Autoimmune Syndromes: Immunogenetics and Long-Term Follow-Up. J. Clin. Endocrinol. Metab. 2003, 88, 2983–2992. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, S.M.; Fallahi, P.; Ruffilli, I.; Elia, G.; Ragusa, F.; Benvenga, S.; Antonelli, A. The association of other autoimmune diseases in patients with Graves’ disease (with or without ophthalmopathy): Review of the literature and report of a large series. Autoimmun. Rev. 2019, 18, 287–292. [Google Scholar] [CrossRef]
- Chang, R.; Chu, K.-A.; Lin, M.-C.; Chu, Y.-H.; Hung, Y.-M.; Wei, J.C. Newly diagnosed iron deficiency anemia and subsequent autoimmune disease: A matched cohort study in Taiwan. Curr. Med Res. Opin. 2020, 36, 985–992. [Google Scholar] [CrossRef]
- Luo, J.; Wang, X.; Yuan, L.; Guo, L. Iron Deficiency, a Risk Factor of Thyroid Disorders in Reproductive-Age and Pregnant Women: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2021, 25, 629831. [Google Scholar] [CrossRef]
- Bach, J.F. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 2002, 347, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, L.; Ramasawmy, R.; Kalil, J. Rheumatic Fever and Rheumatic Heart Disease: Genetics and Pathogenesis. Scand. J. Immunol. 2007, 66, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Yuki, N. Current cases in which epitope mimicry is considered a component cause of autoimmune disease: Guillain-Barré syndrome. Cell Mol. Life Sci. 2000, 57, 527–533. [Google Scholar] [CrossRef]
- Rose, N.R.; Mackay, I.R. Molecular mimicry: A critical look at exemplary instances in human diseases. Cell. Mol. Life Sci. 2000, 57, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Uibo, R. Contribution Of Epidemiological Studies To Gastritis Immunology. Int. Rev. Immunol. 2005, 24, 31–54. [Google Scholar] [CrossRef] [PubMed]
- Jabara, H.H.; Boyden, S.E.; Chou, J.; Ramesh, N.; Massaad, M.J.; Benson, H.; Bainter, W.; Fraulino, D.; Rahimov, F.; Sieff, C.; et al. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat. Genet. 2016, 48, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Hershko, C.; Peto, T.E.; Weatherall, D.J. Iron and infection. Br. Med. J. 1988, 296, 660–664. [Google Scholar] [CrossRef] [Green Version]
- Jason, J.; Archibald, L.K.; Nwanyanwu, O.C.; Bell, M.; Jensen, R.J.; Gunter, E.; Buchanan, I.; Larned, J.; Kazembe, P.N.; Dobbie, H.; et al. The effects of iron deficiency on lymphocyte cytokine production and activation: Preservation of hepatic iron but not at all cost. Clin. Exp. Immunol. 2001, 126, 466–473. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-kappaB Transcription Factors in the Immune System. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Wang, L.; Harrington, L.; Trebicka, E.; Shi, H.N.; Kagan, J.C.; Hong, C.C.; Lin, H.Y.; Babitt, J.L.; Cherayil, B.J. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J. Clin. Investig. 2009, 119, 3322–3328. [Google Scholar] [CrossRef] [Green Version]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar] [CrossRef] [Green Version]
- Gomez, M.A.; Alisaraie, L.; Shio, M.T.; Berghuis, A.M.; Lebrun, C.; Gautier-Luneau, I.; Olivier, M. Protein Tyrosine Phosphatases Are Regulated by Mononuclear Iron Dicitrate. J. Biol. Chem. 2010, 285, 24620–24628. [Google Scholar] [CrossRef] [PubMed]
- Kochanowski, B.A.; Sherman, A.R. Cellular Growth in Iron-Deficient Rats: Effect of Pre- and Postweaning Iron Repletion. J. Nutr. 1985, 115, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Spear, A.T.; Sherman, A.R. Iron Deficiency Alters DMBA-Induced Tumor Burden and Natural Killer Cell Cytotoxicity in Rats. J. Nutr. 1992, 122, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Hassan, T.H.; Badr, M.A.; Karam, N.A.; Zkaria, M.; El Saadany, H.F.; Abdel Rahman, D.M.; Shahbah, D.A.; Al Morshedy, S.M.; Fathy, M.; Esh, A.M.H.; et al. Impact of iron deficiency anemia on the function of the immune system in children. Medicine 2016, 95, e5395. [Google Scholar] [CrossRef]
- Kuvibidila, S.R.; Kitchens, D.; Baliga, B.S. In vivo and in vitro iron deficiency reduces protein kinase C activity and translocation in murine splenic and purified T cells. J. Cell. Biochem. 1999, 74, 468–478. [Google Scholar] [CrossRef]
- Reddy, B.S.; Pleasants, J.R.; Wostmann, B.S. Effect of Intestinal Microflora on Iron and Zinc Metabolism, and on Activities of Metalloenzymes in Rats. J. Nutr. 1972, 102, 101–107. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, C.; Wu, Q.; An, P.; Huang, L.; Wang, J.; Chen, C.; Chen, X.; Zhang, F.; Ma, L.; et al. Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses. Nat. Commun. 2019, 10, 2935. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Obesity and Overweight. 2020. Available online: http://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 16 July 2021).
- International Diabetes Federation. IDF Diabetes Atlas, 1st ed.; International Diabetes Federation: Brussels, Belgium, 2000; Available online: https://www.diabetesatlas.org (accessed on 16 July 2021).
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019; Available online: https://www.diabetesatlas.org (accessed on 16 July 2021).
- Góralska, M.; Majewska-Szczepanik, M.; Szczepanik, M. Mechanizmy immunologiczne towarzyszące otyłości i ich rola w zaburzeniach metabolizmu [Immunological mechanisms involved in obesity and their role in metabolic syndrome]. Postepy Hig. Med. Dosw. 2015, 69, 1384–1404. (In Polish) [Google Scholar]
- Versini, M.; Jeandel, P.-Y.; Rosenthal, E.; Shoenfeld, Y. Obesity in autoimmune diseases: Not a passive bystander. Autoimmun. Rev. 2014, 13, 981–1000. [Google Scholar] [CrossRef]
- Micozzi, M.S.; Albanes, D.; Stevens, R.G. Relation of body size and composition to clinical biochemical and hematologic indices in US men and women. Am. J. Clin. Nutr. 1989, 50, 1276–1281. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D.; Oukka, M.; Koss, L.M.; Aydemir, F.; Wessling-Resnick, M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. USA 2005, 102, 1324–1328. [Google Scholar] [CrossRef] [PubMed]
- Pinhas-Hamiel, O.; Newfield, R.S.; Koren, I.; Agmon, A.; Lilos, P.; Phillip, M. Greater prevalence of iron deficiency in overweight and obese children and adolescents. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Barasch, J.; Mori, K. Cell biology: Iron thievery. Nature 2004, 432, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Plummer, J.; Liu, L.; Byrd, A.; Aschner, M.; Erikson, K.M. The impact of obesity on brain iron levels and α-synuclein expression is regionally dependent. Nutr. Neurosci. 2017, 22, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Feng, Y.; Jia, L.; Li, S.; Li, J.; Wang, Z.; Chen, X.; Du, H. Dietary Iron Modulates Glucose and Lipid Homeostasis in Diabetic Mice. Biol. Trace Element Res. 2018, 189, 194–200. [Google Scholar] [CrossRef]
- Muñoz, M.; Botella-Romero, F.; Gómez-Ramírez, S.; Campos, A.; A García-Erce, J. Iron deficiency and anaemia in bariatric surgical patients: Causes, diagnosis and proper management. Nutr. Hosp. 2010, 24, 640–654. [Google Scholar]
- Manios, Y.; Moschonis, G.; Chrousos, G.P.; Lionis, C.; Mougios, V.; Kantilafti, M.; Tzotzola, V.; Skenderi, K.P.; Petridou, A.; Tsalis, G.; et al. The double burden of obesity and iron deficiency on children and adolescents in Greece: The Healthy Growth Study. J. Hum. Nutr. Diet. 2012, 26, 470–478. [Google Scholar] [CrossRef]
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194. [Google Scholar] [CrossRef]
- Yook, J.-S.; Thomas, S.S.; Toney, A.M.; You, M.; Kim, Y.-C.; Liu, Z.; Lee, J.; Chung, S. Dietary Iron Deficiency Modulates Adipocyte Iron Homeostasis, Adaptive Thermogenesis, and Obesity in C57BL/6 Mice. J. Nutr. 2021, 151, 2967–2975. [Google Scholar] [CrossRef] [PubMed]
- Peirce, V.; Carobbio, S.; Vidal-Puig, A. The different shades of fat. Nature 2014, 510, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Townsend, K.L.; Tseng, Y.-H. Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab. 2014, 25, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yook, J.-S.; You, M.; Kim, Y.; Zhou, M.; Liu, Z.; Kim, Y.-C.; Lee, J.; Chung, S. The thermogenic characteristics of adipocytes are dependent on the regulation of iron homeostasis. J. Biol. Chem. 2021, 296, 100452. [Google Scholar] [CrossRef] [PubMed]
- Lidell, M.E.; Betz, M.J.; Dahlqvist Leinhard, O.; Heglind, M.; Elander, L.; Slawik, M.; Mussack, T.; Nilsson, D.; Romu, T.; Nuutila, P.; et al. Evidence for two types of brown adipose tissue in humans. Nat. Med. 2013, 19, 631–634. [Google Scholar] [CrossRef] [Green Version]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scimè, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koskensalo, K.; Raiko, J.; Saari, T.; Saunavaara, V.; Eskola, O.; Nuutila, P.; Saunavaara, J.; Parkkola, R.; Virtanen, K.A. Human Brown Adipose Tissue Temperature and Fat Fraction Are Related to Its Metabolic Activity. J. Clin. Endocrinol. Metab. 2017, 102, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Ambrosi, J.; González-Crespo, I.; Catalán, V.; Rodríguez, A.; Moncada, R.; Valentí, V.; Romero, S.; Ramírez, B.; Silva, C.; Gil, M.J.; et al. Clinical usefulness of abdominal bioimpedance (ViScan) in the determination of visceral fat and its application in the diagnosis and management of obesity and its comorbidities. Clin. Nutr. 2017, 37, 580–589. [Google Scholar] [CrossRef]
- Deshmukh, A.S.; Peijs, L.; Beaudry, J.L.; Jespersen, N.Z.; Nielsen, C.H.; Ma, T.; Brunner, A.D.; Larsen, T.J.; Bayarri-Olmos, R.; Prabhakar, B.S.; et al. Proteomics-Based Comparative Mapping of the Secretomes of Human Brown and White Adipocytes Reveals EPDR1 as a Novel Batokine. Cell Metab. 2019, 30, 963–975.e7. [Google Scholar] [CrossRef]
- Villarroya, F.; Cereijo, R.; Villarroya, J.; Giralt, M. Brown adipose tissue as a secretory organ. Nat. Rev. Endocrinol. 2017, 13, 26–35. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becerril, S.; Rodríguez, A.; Catalán, V.; Ramírez, B.; Unamuno, X.; Portincasa, P.; Gómez-Ambrosi, J.; Frühbeck, G. Functional Relationship between Leptin and Nitric Oxide in Metabolism. Nutrients 2019, 11, 2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudice, J.; Taylor, J.M. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.P.; Milne, K.J.; Hawke, T.J. Adiponectin—Consideration for its Role in Skeletal Muscle Health. Int. J. Mol. Sci. 2019, 20, 1528. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Becerril, S.; Méndez-Giménez, L.; Ramírez, B.; Sáinz, N.; Catalan, V.; Gómez-Ambrosi, J.; Frühbeck, G. Leptin administration activates irisin-induced myogenesis via nitric oxide-dependent mechanisms, but reduces its effect on subcutaneous fat browning in mice. Int. J. Obes. 2014, 39, 397–407. [Google Scholar] [CrossRef]
- Bal, N.C.; Maurya, S.K.; Pani, S.; Sethy, C.; Banerjee, A.; Das, S.; Patnaik, S.; Kundu, C.N. Mild cold induced thermogenesis: Are BAT and skeletal muscle synergistic partners? Biosci. Rep. 2017, 37, BSR20171087. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Stanford, K.I.; Lynes, M.D.; Takahashi, H.; Baer, L.A.; Arts, P.J.; May, F.J.; Lehnig, A.C.; Middelbeek, R.J.W.; Richard, J.J.; So, K.; et al. 12,13-diHOME: An Exercise-Induced Lipokine that Increases Skeletal Muscle Fatty Acid Uptake. Cell Metab. 2018, 27, 1111–1120, Erratum in Cell. Metab. 2018, 27, 1357. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e12. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, B.; Liang, C.; Li, Y.; Song, Y.-H. Cytokine Signaling in Skeletal Muscle Wasting. Trends Endocrinol. Metab. 2016, 27, 335–347. [Google Scholar] [CrossRef]
- Bonner, J.S.; Lantier, L.; Hasenour, C.M.; James, F.D.; Bracy, D.P.; Wasserman, D.H. Muscle-specific vascular endothelial growth factor deletion induces muscle capillary rarefaction creating muscle insulin resistance. Diabetes 2013, 62, 572–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeremic, N.; Chaturvedi, P.; Tyagi, S.C. Browning of White Fat: Novel Insight Into Factors, Mechanisms, and Therapeutics. J. Cell. Physiol. 2016, 232, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Forner, F.; Kumar, C.; Luber, C.A.; Fromme, T.; Klingenspor, M.; Mann, M. Proteome Differences between Brown and White Fat Mitochondria Reveal Specialized Metabolic Functions. Cell Metab. 2009, 10, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Maretich, P.; Kajimura, S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol. Metab. 2018, 29, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Min, S.Y.; Desai, A.; Yang, Z.; Sharma, A.; DeSouza, T.; Genga, R.M.J.; Kucukural, A.; Lifshitz, L.M.; Nielsen, S.; Scheele, C.; et al. Diverse repertoire of human adipocyte subtypes develops from transcriptionally distinct mesenchymal progenitor cells. Proc. Natl. Acad. Sci. USA 2019, 116, 17970–17979. [Google Scholar] [CrossRef] [Green Version]
- Festa, M.; Ricciardelli, G.; Mele, G.; Pietropaolo, C.; Ruffo, A.; Colonna, A. Overexpression of H Ferritin and Up-regulation of Iron Regulatory Protein Genes during Differentiation of 3T3-L1 Pre-adipocytes. J. Biol. Chem. 2000, 275, 36708–36712. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Barrientos, T.; Andrews, N.C. Iron and Copper in Mitochondrial Diseases. Cell Metab. 2013, 17, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Baynes, R.D.; Bothwell, T.H. Iron Deficiency. Annu. Rev. Nutr. 1990, 10, 133–148. [Google Scholar] [CrossRef]
- Dallman, P.R. Biochemical Basis for the Manifestations of Iron Deficiency. Annu. Rev. Nutr. 1986, 6, 13–40. [Google Scholar] [CrossRef]
- Maguire, J.J.; Davies, K.J.; Dallman, P.R.; Packer, L. Effects of dietary iron deficiency on iron-sulfur proteins and bioenergetic functions of skeletal muscle mitochondria. Biochim. Biophys. Acta 1982, 679, 210–220. [Google Scholar] [CrossRef]
- McKay, R.H.; Higuchi, D.A.; Winder, W.W.; Fell, R.D.; Brown, E.B. Tissue effects of iron deficiency in the rat. Biochim. Biophys. Acta 1983, 757, 352–358. [Google Scholar] [CrossRef]
- Davies, K.J.; Maguire, J.J.; Brooks, G.A.; Dallman, P.R.; Packer, L. Muscle mitochondrial bioenergetics, oxygen supply, and work capacity during dietary iron deficiency and repletion. Am. J. Physiol. 1982, 242, E418–E427. [Google Scholar] [CrossRef] [PubMed]
- Willis, W.T.; Brooks, G.A.; Henderson, S.A.; Dallman, P.R. Effects of iron deficiency and training on mitochondrial enzymes in skeletal muscle. J. Appl. Physiol. 1987, 62, 2442–2446. [Google Scholar] [CrossRef]
- Begriche, K.; Massart, J.; Abbey-Toby, A.; Igoudjil, A.; Lettéron, P.; Fromenty, B. β-Aminoisobutyric Acid Prevents Diet-induced Obesity in Mice With Partial Leptin Deficiency. Obesity 2008, 16, 2053–2067. [Google Scholar] [CrossRef] [PubMed]
- Klempa, K.L.; Willis, W.T.; Chengson, R.; Dallman, P.R.; Brooks, G.A. Iron deficiency decreases gluconeogenesis in isolated rat hepatocytes. J. Appl. Physiol. 1989, 67, 1868–1872. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.L. Iron Biology in Immune Function, Muscle Metabolism and Neuronal Functioning. J. Nutr. 2001, 131, 568S–580S. [Google Scholar] [CrossRef] [Green Version]
- Celsing, F.; Blomstrand, E.; Werner, B.; Pihlstedt, P.; Ekblom, B. Effects of iron deficiency on endurance and muscle enzyme activity in man. Med. Sci. Sports Exerc. 1986, 18, 156–161. [Google Scholar] [CrossRef]
- Edgerton, V.R.; Gardner, G.W.; Ohira, Y.; A Gunawardena, K.; Senewiratne, B. Iron-deficiency anaemia and its effect on worker productivity and activity patterns. BMJ 1979, 2, 1546–1549. [Google Scholar] [CrossRef] [Green Version]
- Perkkio, M.V.; Jansson, L.T.; Brooks, G.A.; Refino, C.J.; Dallman, P.R. Work performance in iron deficiency of increasing severity. J. Appl. Physiol. 1985, 58, 1477–1480. [Google Scholar] [CrossRef]
- Crielaard, B.J.; Lammers, T.; Rivella, S. Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov. 2017, 16, 400–423. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, S.; Raschke, S.; Knebel, B.; Scheler, M.; Irmler, M.; Passlack, W.; Muller, S.; Hanisch, F.-G.; Franz, T.; Li, X.; et al. Secretome profiling of primary human skeletal muscle cells. Biochim. Biophys. Acta 2014, 1844, 1011–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bihan, M.-C.; Bigot, A.; Jensen, S.S.; Dennis, J.L.; Rogowska-Wrzesinska, A.; Lainé, J.; Gache, V.; Furling, D.; Jensen, O.N.; Voit, T.; et al. In-depth analysis of the secretome identifies three major independent secretory pathways in differentiating human myoblasts. J. Proteom. 2012, 77, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J. The hypothalamic–pituitary–adrenal axis in the pathogenesis of rheumatic diseases. Endocrinol. Metab. Clin. N. Am. 2002, 31, 1–13. [Google Scholar] [CrossRef]
- Bilski, J.; Mazur-Bialy, A.I.; Surmiak, M.; Hubalewska-Mazgaj, M.; Pokorski, J.; Nitecki, J.; Nitecka, E.; Pokorska, J.; Targosz, A.; Ptak-Belowska, A.; et al. Effect of Acute Sprint Exercise on Myokines and Food Intake Hormones in Young Healthy Men. Int. J. Mol. Sci. 2020, 21, 8848. [Google Scholar] [CrossRef]
- Mishra, D.; Richard, J.E.; Maric, I.; Porteiro, B.; Häring, M.; Kooijman, S.; Musovic, S.; Eerola, K.; López-Ferreras, L.; Peris, E.; et al. Parabrachial Interleukin-6 Reduces Body Weight and Food Intake and Increases Thermogenesis to Regulate Energy Metabolism. Cell Rep. 2019, 26, 3011–3026.e5. [Google Scholar] [CrossRef] [Green Version]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. Elife 2016, 5, e15092. [Google Scholar] [CrossRef]
- Bartholmey, S.J.; Sherman, A.R. Impaired Ketogenesis in Iron-Deficient Rat Pups. J. Nutr. 1986, 116, 2180–2189. [Google Scholar] [CrossRef]
- Oexle, H.; Gnaiger, E.; Weiss, G. Iron-dependent changes in cellular energy metabolism: Influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta 1999, 1413, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, M. Interleukins and exercise. J. Physiol. 2000, 529 Pt 1, 1. [Google Scholar] [CrossRef]
- Barik, A.; Lu, Y.; Sathyamurthy, A.; Bowman, A.; Shen, C.; Li, L.; Xiong, W.C.; Mei, L. LRP4 is critical for neuromuscular junction maintenance. J. Neurosci. 2014, 34, 13892–13905, Erratum in J. Neurosci. 2015, 35, 7655. [Google Scholar] [CrossRef] [Green Version]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seldin, M.M.; Peterson, J.M.; Byerly, M.S.; Wei, Z.; Wong, G.W. Myonectin (CTRP15), a Novel Myokine That Links Skeletal Muscle to Systemic Lipid Homeostasis. J. Biol. Chem. 2012, 287, 11968–11980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, L.S.; Anderson, B.G.; Strait-Bodey, L.; Stroud, A.M.; Argilés, J.M. Oversecretion of interleukin-15 from skeletal muscle reduces adiposity. Am. J. Physiol. Metab. 2009, 296, E191–E202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dela, F.; von Linstow, M.E.; Mikines, K.J.; Galbo, H. Physical training may enhance β-cell function in type 2 diabetes. Am. J. Physiol. Metab. 2004, 287, E1024–E1031. [Google Scholar] [CrossRef] [PubMed]
- Fiuza-Luces, C.; Garatachea, N.; Berger, N.A.; Lucia, A. Exercise is the Real Polypill. Physiology 2013, 28, 330–358. [Google Scholar] [CrossRef] [Green Version]
- Ellingsgaard, H.; Hauselmann, I.; Schuler, B.; Habib, A.M.; Baggio, L.L.; Meier, D.T.; Eppler, E.; Bouzakri, K.; Wueest, S.; Muller, Y.D.; et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. 2011, 17, 1481–1489. [Google Scholar] [CrossRef] [Green Version]
- Carey, A.L.; Steinberg, G.R.; Macaulay, S.L.; Thomas, W.G.; Holmes, A.G.; Ramm, G.; Prelovsek, O.; Hohnen-Behrens, C.; Watt, M.J.; James, D.E.; et al. Interleukin-6 Increases Insulin-Stimulated Glucose Disposal in Humans and Glucose Uptake and Fatty Acid Oxidation In Vitro via AMP-Activated Protein Kinase. Diabetes 2006, 55, 2688–2697. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Du, F.; Li, X.; Wang, M.; Duan, R.; Zhang, J.; Wu, Y.; Zhang, Q. Effects and underlying mechanisms of irisin on the proliferation and apoptosis of pancreatic β cells. PLoS ONE 2017, 12, e0175498. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.; Neinast, M.; Arany, Z.P. Myobolites: Muscle-derived metabolites with paracrine and systemic effects. Curr. Opin. Pharmacol. 2017, 34, 15–20. [Google Scholar] [CrossRef]
- McCarthy, J.J.; Esser, K.A.; Peterson, C.A.; Dupont-Versteegden, E.E. Evidence of MyomiR network regulation of β-myosin heavy chain gene expression during skeletal muscle atrophy. Physiol. Genom. 2009, 39, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Osmai, M.; Osmai, Y.; Bang-Berthelsen, C.H.; Pallesen, E.M.H.; Vestergaard, A.L.; Novotny, G.W.; Pociot, F.; Mandrup-Poulsen, T. MicroRNAs as regulators of beta-cell function and dysfunction. Diabetes/Metab. Res. Rev. 2015, 32, 334–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forouhi, N.G.; Harding, A.H.; Allison, M.; Sandhu, M.S.; Welch, A.; Luben, R.; Bingham, S.; Khaw, K.T.; Wareham, N.J. Elevated serum ferritin levels predict new-onset type 2 diabetes: Results from the EPIC-Norfolk prospective study. Diabetologia 2007, 50, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Manson, J.E.; Meigs, J.B.; Ma, J.; Rifai, N.; Hu, F.B. Body iron stores in relation to risk of type 2 diabetes in apparently healthy women. JAMA 2004, 291, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, M.-R.; Park, J.-K.; Choi, W.-J.; Cho, A.-R.; Lee, Y.-J. Serum ferritin level is positively associated with insulin resistance and metabolic syndrome in postmenopausal women: A nationwide population-based study. Maturitas 2017, 103, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Yokomori, N.; Iwasa, Y.; Aida, K.; Inoue, M.; Tawata, M.; Onaya, T. Transcriptional Regulation of Ferritin Messenger Ribonucleic Acid Levels by Insulin in Cultured Rat Glioma Cells. Endocrinology 1991, 128, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, L.J.; Mittelman, S.D.; Watanabe, R.M.; Coates, T.D.; Wood, J.C. Pancreatic iron and glucose dysregulation in thalassemia major. Am. J. Hematol. 2011, 87, 155–160. [Google Scholar] [CrossRef]
- Fleming, R.E.; Sly, W.S. Mechanisms of Iron Accumulation in Hereditary Hemochromatosis. Annu. Rev. Physiol. 2002, 64, 663–680. [Google Scholar] [CrossRef]
- Fernández-Real, J.M.; Moreno, J.M.; López-Bermejo, A.; Chico, B.; Vendrell, J.; Ricart, W. Circulating Soluble Transferrin Receptor According to Glucose Tolerance Status and Insulin Sensitivity. Diabetes Care 2007, 30, 604–608. [Google Scholar] [CrossRef] [Green Version]
- Koch, R.O.; Zoller, H.; Theuri, I.; Obrist, P.; Egg, G.; Strohmayer, W.; Vogel, W.; Weiss, G. Distribution of DMT 1 within the human glandular system. Histol. Histopathol. 2003, 18, 1095–1101. [Google Scholar] [CrossRef]
- Summers, K.L.; Fimognari, N.; Hollings, A.; Kiernan, M.; Lam, V.; Tidy, R.J.; Paterson, D.; Tobin, M.J.; Takechi, R.; George, G.N.; et al. A Multimodal Spectroscopic Imaging Method To Characterize the Metal and Macromolecular Content of Proteinaceous Aggregates (“Amyloid Plaques”). Biochemistry 2017, 56, 4107–4116. [Google Scholar] [CrossRef] [Green Version]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Paquot, N.; Scheen, A.J. Inflammasome NLRP3 et graisse viscérale [NLRP3 inflammasome and visceral adipose tissue]. Rev. Med. Liege 2014, 69, 57–61. (In French) [Google Scholar] [PubMed]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin, cognition, and dementia. Eur. J. Pharmacol. 2013, 719, 170–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohira, Y.; Chen, C.-S.; Hegenauer, J.; Saltman, P. Adaptations of Lactate Metabolism in Iron-Deficient Rats. Exp. Biol. Med. 1983, 173, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.H.; Green, Y.S.; Ledingham, J.G.; Radda, G.K.; Rajagopalan, B. The effect of iron deficiency on skeletal muscle metabolism of the rat. Acta Physiol. Scand. 1993, 147, 85–90. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Schaible, U.E.; Kaufmann, S.H. Iron and microbial infection. Nat. Rev. Microbiol. 2004, 2, 946–953, Erratum in Nat. Rev. Microbiol. 2005, 3, 268. [Google Scholar] [CrossRef]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and Propionate Protect against Diet-Induced Obesity and Regulate Gut Hormones via Free Fatty Acid Receptor 3-Independent Mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [Green Version]
- Bellahcene, M.; O’Dowd, J.F.; Wargent, E.T.; Zaibi, M.S.; Hislop, D.C.; Ngala, R.A.; Smith, D.M.; Cawthorne, M.A.; Stocker, C.J.; Arch, J.R.S. Male mice that lack the G-protein-coupled receptor GPR41 have low energy expenditure and increased body fat content. Br. J. Nutr. 2012, 109, 1755–1764. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.; Suh, J.M.; Reilly, S.M.; Yu, E.; Osborn, O.; Lackey, D.; Yoshihara, E.; Perino, A.; Jacinto, S.; Lukasheva, Y.; et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 2015, 21, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Shorb, S.R. Anemia and Diabetic Retinopathy. Am. J. Ophthalmol. 1985, 100, 434–436. [Google Scholar] [CrossRef]
- Robles, N.R.; Ramos, J.L.; Chavez, E.; Gonzalez Candia, B.; Bayo, M.A.; Cidoncha, A.; Gomez, J.L.; Cubero, J.J. Iron deficiency in chronic kidney disease patients with diabetes mellitus. Diabetes Metab. Syndr. 2018, 12, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Praveen, M.; Jain, N.; Raizada, N.; Sharma, S.; Narang, S.; Madhu, S. Anaemia in patients with type 2 diabetes mellitus without nephropathy is related to iron deficiency. Diabetes Metab. Syndr. 2020, 14, 1837–1840. [Google Scholar] [CrossRef] [PubMed]
- Stoebner, R.; Kiser, R.; Alperin, J.B. Iron deficiency anemia and papilledema. Rapid resolution with oral iron therapy. Am. J. Dig. Dis. 1970, 15, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Bergis, D.; Tessmer, L.; Badenhoop, K. Iron deficiency in long standing type 1 diabetes mellitus and its association with depression and impaired quality of life. Diabetes Res. Clin. Pract. 2019, 151, 74–81. [Google Scholar] [CrossRef]
- Breining, P.; Jensen, J.B.; Sundelin, E.I.; Gormsen, L.C.; Jakobsen, S.; Busk, M.; Rolighed, L.; Bross, P.; Fernandez-Guerra, P.; Markussen, L.K.; et al. Metformin targets brown adipose tissue in vivo and reduces oxygen consumption in vitro. Diabetes Obes. Metab. 2018, 20, 2264–2273. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szklarz, M.; Gontarz-Nowak, K.; Matuszewski, W.; Bandurska-Stankiewicz, E. “Ferrocrinology”—Iron Is an Important Factor Involved in Gluco- and Lipocrinology. Nutrients 2022, 14, 4693. https://doi.org/10.3390/nu14214693
Szklarz M, Gontarz-Nowak K, Matuszewski W, Bandurska-Stankiewicz E. “Ferrocrinology”—Iron Is an Important Factor Involved in Gluco- and Lipocrinology. Nutrients. 2022; 14(21):4693. https://doi.org/10.3390/nu14214693
Chicago/Turabian StyleSzklarz, Michał, Katarzyna Gontarz-Nowak, Wojciech Matuszewski, and Elżbieta Bandurska-Stankiewicz. 2022. "“Ferrocrinology”—Iron Is an Important Factor Involved in Gluco- and Lipocrinology" Nutrients 14, no. 21: 4693. https://doi.org/10.3390/nu14214693