
High Folate, Perturbed One-Carbon Metabolism and Gestational Diabetes Mellitus
 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Gestational Diabetes Mellitus (GDM): Health Significance
3. Gestational Diabetes Mellitus: Risk Factors and the Role of Folic Acid in GDM Aetiology
3.1. High FA and GDM Risk
3.2. High FA and Metabolic Dysfunction
3.3. High FA and β-Cell Dysfunction
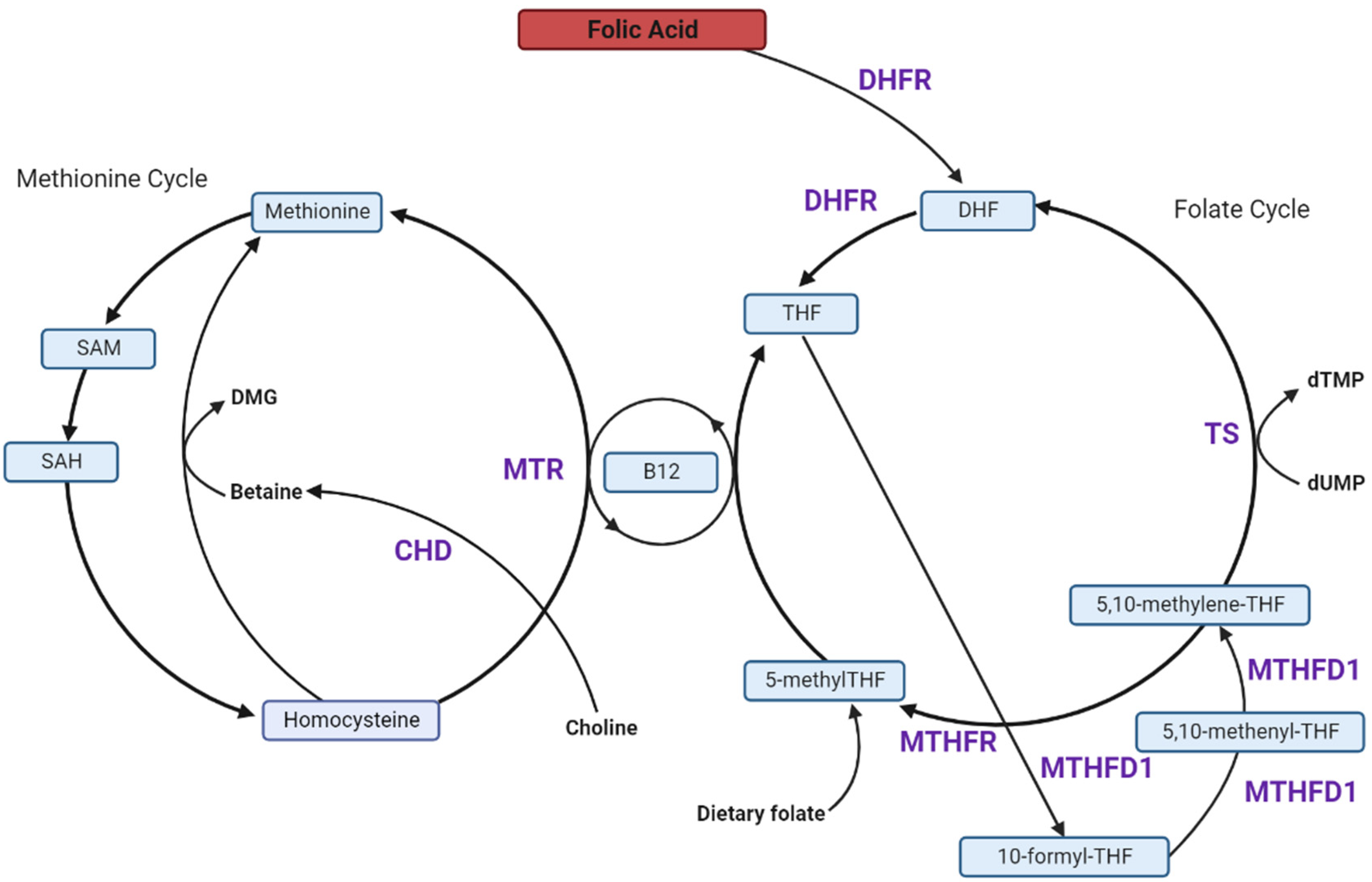
4. Folate and One-Carbon Metabolism in Pregnancy
5. High FA Intake and Unmetabolized FA (uFA)
6. Effects of Excess FA on the Players in the 1C-Metabolism Pathway
6.1. Excess FA Saturates Limited DHFR Capacity
6.2. Excess FA Reduces MTHFR Protein, Causing a Pseudo-MTHFR Deficiency
6.3. FA Reduces Methionine Synthase (MTR) Activity and Favours the Thymidylate Synthase (TS) Cycle
6.4. Dietary FA Alters Choline and Betaine Metabolism
7. Association between One-Carbon Metabolism Players and GDM
7.1. High FA and Low Vitamin B12 Are Associated with Increased GDM Risk
7.2. Circulating Homocysteine Is Elevated in GDM-Complicated Pregnancies
7.3. Interactions between Excess Folate, Vitamin B12, Homocysteine and Risk for GDM
7.4. Choline-Derived Betaine Is Associated with Decreased GDM Risk
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiefari, E.; Arcidiacono, B.; Foti, D.; Brunetti, A. Gestational diabetes mellitus: An updated overview. J. Endocrinol. Investig. 2017, 40, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Australian Institute of Health and Welfare. Incidence of Gestational Diabetes in Australia, Cat. No: CVD 85; AIHW: Canberra, Australia, 2019. [Google Scholar]
- Australian Institute of Health and Welfare. Diabetes, Cat. No: CVD 82; AIHW: Canberra, Australia, 2020. [Google Scholar]
- Palchetti, C.Z.; Paniz, C.; de Carli, E.; Marchioni, D.M.; Colli, C.; Steluti, J.; Pfeiffer, C.M.; Fazili, Z.; Guerra-Shinohara, E.M. Association between Serum Unmetabolized Folic Acid Concentrations and Folic Acid from Fortified Foods. J. Am. Coll. Nutr. 2017, 36, 572–578. [Google Scholar] [CrossRef]
- Wright, A.J.A.; Dainty, J.R.; Finglas, P.M. Folic acid metabolism in human subjects revisited: Potential implications for proposed mandatory folic acid fortification in the UK. Br. J. Nutr. 2007, 98, 667–675. [Google Scholar] [CrossRef]
- Pfeiffer, C.M.; Sternberg, M.R.; Fazili, Z.; Yetley, E.A.; Lacher, D.A.; Bailey, R.L.; Johnson, C.L. Unmetabolized folic acid is detected in nearly all serum samples from US children, adolescents, and adults. J. Nutr. 2015, 145, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Ge, X.; Huang, K.; Mao, L.; Yan, S.; Xu, Y.; Huang, S.; Hao, J.; Zhu, P.; Niu, Y.; et al. Folic Acid Supplement Intake in Early Pregnancy Increases Risk of Gestational Diabetes Mellitus: Evidence from a Prospective Cohort Study. Diabetes Care 2016, 39, e36–e37. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Y.; Chen, H.; Jiang, Y.; Wang, Y.; Wang, D.; Li, M.; Dou, Y.; Sun, X.; Huang, G.; et al. Association of Maternal Folate and Vitamin B12 in Early Pregnancy with Gestational Diabetes Mellitus: A Prospective Cohort Study. Diabetes Care 2021, 44, 217–223. [Google Scholar] [CrossRef]
- Cheng, G.; Sha, T.; Gao, X.; He, Q.; Wu, X.; Tian, Q.; Yang, F.; Tang, C.; Wu, X.; Xie, Q.; et al. The Associations between the Duration of Folic Acid Supplementation, Gestational Diabetes Mellitus, and Adverse Birth Outcomes based on a Birth Cohort. Int. J. Environ. Res. Public Health 2019, 16, 4511. [Google Scholar] [CrossRef]
- Huang, L.; Yu, X.; Li, L.; Chen, Y.; Yang, Y.; Yang, Y.; Hu, Y.; Zhao, Y.; Tang, H.; Xu, D.; et al. Duration of periconceptional folic acid supplementation and risk of gestational diabetes mellitus. Asia Pac. J. Clin. Nutr. 2019, 28, 321–329. [Google Scholar] [CrossRef]
- Xie, K.; Xu, P.; Fu, Z.; Gu, X.; Li, H.; Cui, X.; You, L.; Zhu, L.; Ji, C.; Guo, X. Association of maternal folate status in the second trimester of pregnancy with the risk of gestational diabetes mellitus. Food Sci. Nutr. 2019, 7, 3759–3765. [Google Scholar] [CrossRef]
- Liu, P.J.; Liu, Y.; Ma, L.; Yao, A.M.; Chen, X.Y.; Hou, Y.X.; Wu, L.P.; Xia, L.Y. Associations Between Gestational Diabetes Mellitus Risk and Folate Status in Early Pregnancy and MTHFR C677T Polymorphisms in Chinese Women. Diabetes Metab. Syndr. Obes. 2020, 13, 1499–1507. [Google Scholar] [CrossRef]
- Li, N.; Jiang, J.; Guo, L. Effects of maternal folate and vitamin B12 on gestational diabetes mellitus: A dose-response meta-analysis of observational studies. Eur. J. Clin. Nutr. 2022; in press. [Google Scholar] [CrossRef]
- Li, S.; Hou, Y.; Yan, X.; Wang, Y.; Shi, C.; Wu, X.; Liu, H.; Zhang, L.; Zhang, X.; Liu, J.; et al. Joint effects of folate and vitamin B(12) imbalance with maternal characteristics on gestational diabetes mellitus. J. Diabetes 2019, 11, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, P.; Sukumar, N.; Adaikalakoteswari, A.; Goljan, I.; Venkataraman, H.; Gopinath, A.; Bagias, C.; Yajnik, C.S.; Stallard, N.; Ghebremichael-Weldeselassie, Y.; et al. Association of maternal vitamin B12 and folate levels in early pregnancy with gestational diabetes: A prospective UK cohort study (PRiDE study). Diabetologia 2021, 64, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Jankovic-Karasoulos, T.; Furness, D.L.; Leemaqz, S.Y.; Dekker, G.A.; Grzeskowiak, L.E.; Grieger, J.A.; Andraweera, P.H.; McCullough, D.; McAninch, D.; McCowan, L.M.; et al. Maternal folate, one-carbon metabolism and pregnancy outcomes. Matern. Child Nutr. 2021, 17, e13064. [Google Scholar] [CrossRef] [PubMed]
- Lowe, W.L., Jr.; Scholtens, D.M.; Lowe, L.P.; Kuang, A.; Nodzenski, M.; Talbot, O.; Catalano, P.M.; Linder, B.; Brickman, W.J.; Clayton, P.; et al. Association of Gestational Diabetes with Maternal Disorders of Glucose Metabolism and Childhood Adiposity. JAMA 2018, 320, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Metzger, B.E.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; McIntyre, H.D.; et al. Hyperglycemia and adverse pregnancy outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, W.; Leng, J.; Zhang, S.; Liu, H.; Li, W.; Wang, L.; Tian, H.; Chen, J.; Qi, L.; et al. High risk of metabolic syndrome after delivery in pregnancies complicated by gestational diabetes. Diabetes Res. Clin. Pract. 2019, 150, 219–226. [Google Scholar] [CrossRef]
- Kaiser, K.; Nielsen, M.F.; Kallfa, E.; Dubietyte, G.; Lauszus, F.F. Metabolic syndrome in women with previous gestational diabetes. Sci. Rep. 2021, 11, 11558. [Google Scholar] [CrossRef] [PubMed]
- Pathirana, M.M.; Lassi, Z.S.; Ali, A.; Arstall, M.A.; Roberts, C.T.; Andraweera, P.H. Association between metabolic syndrome and gestational diabetes mellitus in women and their children: A systematic review and meta-analysis. Endocrine 2021, 71, 310–320. [Google Scholar] [CrossRef]
- Bellamy, L.; Casas, J.-P.; Hingorani, A.D.; Williams, D. Type 2 diabetes mellitus after gestational diabetes: A systematic review and meta-analysis. Lancet 2009, 373, 1773–1779. [Google Scholar] [CrossRef]
- Goueslard, K.; Cottenet, J.; Mariet, A.-S.; Giroud, M.; Cottin, Y.; Petit, J.-M.; Quantin, C. Early cardiovascular events in women with a history of gestational diabetes mellitus. Cardiovasc. Diabetol. 2016, 15, 15. [Google Scholar] [CrossRef]
- Kessous, R.; Shoham-Vardi, I.; Pariente, G.; Sherf, M.; Sheiner, E. An association between gestational diabetes mellitus and long-term maternal cardiovascular morbidity. Heart 2013, 99, 1118. [Google Scholar] [CrossRef]
- Pathirana, M.M.; Lassi, Z.; Ali, A.; Arstall, M.; Roberts, C.T.; Andraweera, P.H. Cardiovascular risk factors in women with previous gestational diabetes mellitus: A systematic review and meta-analysis. Rev. Endocr. Metab. Disord. 2021, 22, 729–761. [Google Scholar] [CrossRef] [PubMed]
- Pathirana, M.M.; Lassi, Z.S.; Roberts, C.T.; Andraweera, P.H. Cardiovascular risk factors in offspring exposed to gestational diabetes mellitus in utero: Systematic review and meta-analysis. J. Dev. Orig. Health Dis. 2020, 11, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Abokaf, H.; Shoham-Vardi, I.; Sergienko, R.; Landau, D.; Sheiner, E. In utero exposure to gestational diabetes mellitus and long term endocrine morbidity of the offspring. Diabetes Res. Clin. Pract. 2018, 144, 231–235. [Google Scholar] [CrossRef]
- Bush, N.C.; Chandler-Laney, P.C.; Rouse, D.J.; Granger, W.M.; Oster, R.A.; Gower, B.A. Higher maternal gestational glucose concentration is associated with lower offspring insulin sensitivity and altered beta-cell function. J. Clin. Endocrinol. Metab. 2011, 96, E803–E809. [Google Scholar] [CrossRef]
- Kelstrup, L.; Damm, P.; Mathiesen, E.R.; Hansen, T.; Vaag, A.A.; Pedersen, O.; Clausen, T.D. Insulin Resistance and Impaired Pancreatic β-Cell Function in Adult Offspring of Women with Diabetes in Pregnancy. J. Clin. Endocrinol. Metab. 2013, 98, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Lean, S.C.; Derricott, H.; Jones, R.L.; Heazell, A.E.P. Advanced maternal age and adverse pregnancy outcomes: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0186287. [Google Scholar] [CrossRef]
- Giannakou, K.; Evangelou, E.; Yiallouros, P.; Christophi, C.A.; Middleton, N.; Papatheodorou, E.; Papatheodorou, S.I. Risk factors for gestational diabetes: An umbrella review of meta-analyses of observational studies. PLoS ONE 2019, 14, e0215372. [Google Scholar] [CrossRef]
- Caughey, A.B.; Cheng, Y.W.; Stotland, N.E.; Washington, A.E.; Escobar, G.J. Maternal and paternal race/ethnicity are both associated with gestational diabetes. Am. J. Obstet. Gynecol. 2010, 202, 616.e1–616.e5. [Google Scholar] [CrossRef]
- Australian Institute of Health and Welfare. Australia’s Mothers and Babies 2018—In Brief, Cat. No: PER 108; AIHW: Canberra, Australia, 2020. [Google Scholar]
- Australian Institute of Health and Welfare. Australia’s Mothers and Babies, Cat. No: PER 101; AIHW: Canberra, Australia, 2021. [Google Scholar]
- Metzger, B.E.; Gabbe, S.G.; Persson, B.; Buchanan, T.A.; Catalano, P.A.; Damm, P.; Dyer, A.R.; Leiva, A.; Hod, M.; Kitzmiler, J.L.; et al. International association of diabetes and pregnancy study groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care 2010, 33, 676–682. [Google Scholar] [CrossRef]
- Krishnaveni, G.V.; Veena, S.R.; Karat, S.C.; Yajnik, C.S.; Fall, C.H.D. Association between maternal folate concentrations during pregnancy and insulin resistance in Indian children. Diabetologia 2014, 57, 110–121. [Google Scholar] [CrossRef]
- Yajnik, C.S.; Deshpande, S.S.; Jackson, A.A.; Refsum, H.; Rao, S.; Fisher, D.J.; Bhat, D.S.; Naik, S.S.; Coyaji, K.J.; Joglekar, C.V.; et al. Vitamin B12 and folate concentrations during pregnancy and insulin resistance in the offspring: The Pune Maternal Nutrition Study. Diabetologia 2008, 51, 29–38. [Google Scholar] [CrossRef]
- Tojal, A.; Neves, C.; Veiga, H.; Ferreira, S.; Rodrigues, I.; Martel, F.; Calhau, C.; Negrão, R.; Keating, E. Perigestational high folic acid: Impact on offspring’s peripheral metabolic response. Food Funct. 2019, 10, 7216–7226. [Google Scholar] [CrossRef]
- Huang, Y.; He, Y.; Sun, X.; He, Y.; Li, Y.; Sun, C. Maternal High Folic Acid Supplement Promotes Glucose Intolerance and Insulin Resistance in Male Mouse Offspring Fed a High-Fat Diet. Int. J. Mol. Sci. 2014, 15, 6298–6313. [Google Scholar] [CrossRef] [PubMed]
- Kintaka, Y.; Wada, N.; Shioda, S.; Nakamura, S.; Yamazaki, Y.; Mochizuki, K. Excessive folic acid supplementation in pregnant mice impairs insulin secretion and induces the expression of genes associated with fatty liver in their offspring. Heliyon 2020, 6, e03597. [Google Scholar] [CrossRef]
- Yang, X.; Huang, Y.; Sun, C.; Li, J. Maternal Prenatal Folic Acid Supplementation Programs Offspring Lipid Metabolism by Aberrant DNA Methylation in Hepatic ATGL and Adipose LPL in Rats. Nutrients 2017, 9, 935. [Google Scholar] [CrossRef]
- Cuthbert, C.E.; Foster, J.E.; Ramdath, D.D. A maternal high-fat, high-sucrose diet alters insulin sensitivity and expression of insulin signalling and lipid metabolism genes and proteins in male rat offspring: Effect of folic acid supplementation. Br. J. Nutr. 2017, 118, 580–588. [Google Scholar] [CrossRef]
- Kelly, K.B.; Kennelly, J.P.; Ordonez, M.; Nelson, R.; Leonard, K.; Stabler, S.; Gomez-Muñoz, A.; Field, C.J.; Jacobs, R.L. Excess Folic Acid Increases Lipid Storage, Weight Gain, and Adipose Tissue Inflammation in High Fat Diet-Fed Rats. Nutrients 2016, 8, 594. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Qin, D.; Xu, S.; He, C.; Sun, J.; Hua, J.; Peng, S. Folic acid promotes proliferation and differentiation of porcine pancreatic stem cells into insulin-secreting cells through canonical Wnt and ERK signaling pathway. J. Steroid Biochem. Mol. Biol. 2021, 205, 105772. [Google Scholar] [CrossRef]
- Karampelias, C.; Rezanejad, H.; Rosko, M.; Duan, L.; Lu, J.; Pazzagli, L.; Bertolino, P.; Cesta, C.E.; Liu, X.; Korbutt, G.S.; et al. Reinforcing one-carbon metabolism via folic acid/Folr1 promotes β-cell differentiation. Nat. Commun. 2021, 12, 3362. [Google Scholar] [CrossRef]
- Baeyens, L.; Hindi, S.; Sorenson, R.L.; German, M.S. β-Cell adaptation in pregnancy. Diabetes Obes. Metab. 2016, 18 (Suppl. S1), 63–70. [Google Scholar] [CrossRef]
- Butler, A.E.; Cao-Minh, L.; Galasso, R.; Rizza, R.A.; Corradin, A.; Cobelli, C.; Butler, P.C. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010, 53, 2167–2176. [Google Scholar] [CrossRef]
- Moyce, B.L.; Dolinsky, V.W. Maternal β-Cell Adaptations in Pregnancy and Placental Signalling: Implications for Gestational Diabetes. Int. J. Mol. Sci. 2018, 19, 3467. [Google Scholar] [CrossRef] [PubMed]
- Shulpekova, Y.; Nechaev, V.; Kardasheva, S.; Sedova, A.; Kurbatova, A.; Bueverova, E.; Kopylov, A.; Malsagova, K.; Dlamini, J.C.; Ivashkin, V. The Concept of Folic Acid in Health and Disease. Molecules 2021, 26, 3731. [Google Scholar] [CrossRef]
- Selhub, J. Folate, vitamin B12 and vitamin B6 and one carbon metabolism. J. Nutr. Health Aging 2002, 6, 39–42. [Google Scholar]
- Dai, C.; Fei, Y.; Li, J.; Shi, Y.; Yang, X. A Novel Review of Homocysteine and Pregnancy Complications. BioMed Res. Int. 2021, 2021, 6652231. [Google Scholar] [CrossRef]
- Kalhan, S.C. One carbon metabolism in pregnancy: Impact on maternal, fetal and neonatal health. Mol. Cell. Endocrinol. 2016, 435, 48–60. [Google Scholar] [CrossRef]
- Lan, X.; Field, M.S.; Stover, P.J. Cell cycle regulation of folate-mediated one-carbon metabolism. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1426. [Google Scholar] [CrossRef]
- Crider, K.S.; Yang, T.P.; Berry, R.J.; Bailey, L.B. Folate and DNA methylation: A review of molecular mechanisms and the evidence for folate’s role. Adv. Nutr. 2012, 3, 21–38. [Google Scholar] [CrossRef]
- Korsmo, H.W.; Jiang, X. One carbon metabolism and early development: A diet-dependent destiny. Trends Endocrinol. Metab. 2021, 32, 579–593. [Google Scholar] [CrossRef]
- Scaglione, F.; Panzavolta, G. Folate, folic acid and 5-methyltetrahydrofolate are not the same thing. Xenobiotica 2014, 44, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Beal, T.; Ortenzi, F. Priority Micronutrient Density in Foods. Front. Nutr. 2022, 9, 806566. [Google Scholar] [CrossRef] [PubMed]
- Darcy-Vrillon, B.; Selhub, J.; Rosenberg, I.H. Analysis of sequential events in intestinal absorption of folylpolyglutamate. Am. J. Physiol. Gastrointest. Liver Physiol. 1988, 255, G361–G366. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.N.; Kaldis, P. Pairing structural reconstruction with catalytic competence to evaluate the mechanisms of key enzymes in the folate-mediated one-carbon pathway. FEBS J. 2022; in press. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Goh, Y.I.; Koren, G. Folic acid in pregnancy and fetal outcomes. J. Obstet. Gynaecol. 2008, 28, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.C.; Hayes, D.J.; Jones, R.L. Effects of micronutrients on placental function: Evidence from clinical studies to animal models. Reproduction 2018, 156, R69–R82. [Google Scholar] [CrossRef] [PubMed]
- Osgood, E.E. Development and growth of hematopoietic tissues; with a clinically practical method of growth analysis. Pediatrics 1955, 15, 733–751. [Google Scholar] [CrossRef]
- Hirsch, H.R. The dynamics of repetitive asymmetric cell division. Mech. Ageing Dev. 1977, 6, 319–332. [Google Scholar] [CrossRef]
- Antony, A.C. In utero physiology: Role of folic acid in nutrient delivery and fetal development. Am. J. Clin. Nutr. 2007, 85, 598S–603S. [Google Scholar] [CrossRef]
- Fekete, K.; Berti, C.; Cetin, I.; Hermoso, M.; Koletzko, B.V.; Decsi, T. Perinatal folate supply: Relevance in health outcome parameters. Matern. Child Nutr. 2010, 6, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Picciano, M.F. Folate and human reproduction. Am. J. Clin. Nutr. 2006, 83, 993–1016. [Google Scholar] [CrossRef] [PubMed]
- Bulloch, R.E.; Wall, C.R.; Thompson, J.M.D.; Taylor, R.S.; Poston, L.; Roberts, C.T.; Dekker, G.A.; Kenny, L.C.; Simpson, N.A.B.; Myers, J.E.; et al. Folic acid supplementation is associated with size at birth in the Screening for Pregnancy Endpoints (SCOPE) international prospective cohort study. Early Hum. Dev. 2020, 147, 105058. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, R.; Malone, F.D.; Porter, F.T.; Nyberg, D.A.; Comstock, C.H.; Hankins, G.D.; Eddleman, K.; Gross, S.J.; Dugoff, L.; Craigo, S.D.; et al. Preconceptional folate supplementation and the risk of spontaneous preterm birth: A cohort study. PLoS Med. 2009, 6, e1000061. [Google Scholar] [CrossRef]
- Bulloch, R.E.; Lovell, A.L.; Jordan, V.M.B.; McCowan, L.M.E.; Thompson, J.M.D.; Wall, C.R. Maternal folic acid supplementation for the prevention of preeclampsia: A systematic review and meta-analysis. Paediatr. Perinat. Epidemiol. 2018, 32, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Caro, R.; Fast, J. Pregnancy Myths and Practical Tips. Am. Fam. Physician 2020, 102, 420–426. [Google Scholar]
- Berti, C.; Biesalski, H.K.; Gärtner, R.; Lapillonne, A.; Pietrzik, K.; Poston, L.; Redman, C.; Koletzko, B.; Cetin, I. Micronutrients in pregnancy: Current knowledge and unresolved questions. Clin. Nutr. 2011, 30, 689–701. [Google Scholar] [CrossRef]
- Wilson, R.D.; O’Connor, D.L. Guideline No. 427: Folic Acid and Multivitamin Supplementation for Prevention of Folic Acid-Sensitive Congenital Anomalies. J. Obstet. Gynaecol. Can. 2022, 44, 707–719.e1. [Google Scholar] [CrossRef]
- Wilson, R.D.; O’Connor, D.L. Maternal folic acid and multivitamin supplementation: International clinical evidence with considerations for the prevention of folate-sensitive birth defects. Prev. Med. Rep. 2021, 24, 101617. [Google Scholar] [CrossRef]
- Ledowsky, C.; Mahimbo, A.; Scarf, V.; Steel, A. Women Taking a Folic Acid Supplement in Countries with Mandatory Food Fortification Programs May Be Exceeding the Upper Tolerable Limit of Folic Acid: A Systematic Review. Nutrients 2022, 14, 2715. [Google Scholar] [CrossRef]
- Gernand, A.D.; Schulze, K.J.; Stewart, C.P.; West, K.P.; Christian, P. Micronutrient deficiencies in pregnancy worldwide: Health effects and prevention. Nat. Rev. Endocrinol. 2016, 12, 274–289. [Google Scholar] [CrossRef]
- Mousa, A.; Naqash, A.; Lim, S. Macronutrient and Micronutrient Intake during Pregnancy: An Overview of Recent Evidence. Nutrients 2019, 11, 443. [Google Scholar] [CrossRef]
- Belkacemi, L.; Nelson, D.M.; Desai, M.; Ross, M.G. Maternal Undernutrition Influences Placental-Fetal Development1. Biol. Reprod. 2010, 83, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.S.Q.; Muldoon, K.A.; Sheyholislami, H.; Behan, N.; Lamers, Y.; Rybak, N.; White, R.R.; Harvey, A.L.J.; Gaudet, L.M.; Smith, G.N.; et al. Impact of high-dose folic acid supplementation in pregnancy on biomarkers of folate status and 1-carbon metabolism: An ancillary study of the Folic Acid Clinical Trial (FACT). Am. J. Clin. Nutr. 2021, 113, 1361–1371. [Google Scholar] [CrossRef]
- Plumptre, L.; Masih, S.P.; Ly, A.; Aufreiter, S.; Sohn, K.-J.; Croxford, R.; Lausman, A.Y.; Berger, H.; O’Connor, D.L.; Kim, Y.-I. High concentrations of folate and unmetabolized folic acid in a cohort of pregnant Canadian women and umbilical cord blood. Am. J. Clin. Nutr. 2015, 102, 848–857. [Google Scholar] [CrossRef]
- Sweeney, M.R.; McPartlin, J.; Weir, D.G.; Daly, S.; Pentieva, K.; Daly, L.; Scott, J.M. Evidence of unmetabolised folic acid in cord blood of newborn and serum of 4-day-old infants. Br. J. Nutr. 2005, 94, 727–730. [Google Scholar] [CrossRef]
- Raghavan, R.; Selhub, J.; Paul, L.; Ji, Y.; Wang, G.; Hong, X.; Zuckerman, B.; Fallin, M.D.; Wang, X. A prospective birth cohort study on cord blood folate subtypes and risk of autism spectrum disorder. Am. J. Clin. Nutr. 2020, 112, 1304–1317. [Google Scholar] [CrossRef]
- McGowan, E.C.; Hong, X.; Selhub, J.; Paul, L.; Wood, R.A.; Matsui, E.C.; Keet, C.A.; Wang, X. Association between Folate Metabolites and the Development of Food Allergy in Children. J. Allergy Clin. Immunol. Pract. 2020, 8, 132–140.e5. [Google Scholar] [CrossRef]
- Page, R.; Robichaud, A.; Arbuckle, T.E.; Fraser, W.D.; MacFarlane, A.J. Total folate and unmetabolized folic acid in the breast milk of a cross-section of Canadian women. Am. J. Clin. Nutr. 2017, 105, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Page, R.; Wong, A.; Arbuckle, T.E.; MacFarlane, A.J. The MTHFR 677C>T polymorphism is associated with unmetabolized folic acid in breast milk in a cohort of Canadian women. Am. J. Clin. Nutr. 2019, 110, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Houghton, L.A.; Yang, J.; O’Connor, D.L. Unmetabolized folic acid and total folate concentrations in breast milk are unaffected by low-dose folate supplements. Am. J. Clin. Nutr. 2009, 89, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.R.; Staines, A.; Daly, L.; Traynor, A.; Daly, S.; Bailey, S.W.; Alverson, P.B.; Ayling, J.E.; Scott, J.M. Persistent circulating unmetabolised folic acid in a setting of liberal voluntary folic acid fortification. Implications for further mandatory fortification? BMC Public Health 2009, 9, 295. [Google Scholar] [CrossRef] [PubMed]
- Best, K.P.; Green, T.J.; Sulistyoningrum, D.C.; Sullivan, T.R.; Aufreiter, S.; Prescott, S.L.; Makrides, M.; Skubisz, M.; O’Connor, D.L.; Palmer, D.J. Maternal Late-Pregnancy Serum Unmetabolized Folic Acid Concentrations Are not Associated with Infant Allergic Disease: A Prospective Cohort Study. J. Nutr. 2021, 151, 1553–1560. [Google Scholar] [CrossRef]
- Husebye, E.S.N.; Wendel, A.W.K.; Gilhus, N.E.; Riedel, B.; Bjørk, M.H. Plasma unmetabolized folic acid in pregnancy and risk of autistic traits and language impairment in antiseizure medication-exposed children of women with epilepsy. Am. J. Clin. Nutr. 2022, 115, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Patanwala, I.; King, M.J.; Barrett, D.A.; Rose, J.; Jackson, R.; Hudson, M.; Philo, M.; Dainty, J.R.; Wright, A.J.; Finglas, P.M.; et al. Folic acid handling by the human gut: Implications for food fortification and supplementation. Am. J. Clin. Nutr. 2014, 100, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, B.; Sahyoun, N.R. Application of the Key Events Dose-response Framework to Folate Metabolism. Crit. Rev. Food Sci. Nutr. 2016, 56, 1325–1333. [Google Scholar] [CrossRef]
- Bailey, S.W.; Ayling, J.E. The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proc. Natl. Acad. Sci. USA 2009, 106, 15424–15429. [Google Scholar] [CrossRef]
- Blakley, R.L.; Benkovic, S.J.; Whitehead, V.M. Folates and Pterins, Chemistry and Biochemistry of Folates; Wiley-Interscience: Hoboken, NJ, USA, 1984; Volume 1. [Google Scholar]
- Kelly, P.; McPartlin, J.; Goggins, M.; Weir, D.G.; Scott, J.M. Unmetabolized folic acid in serum: Acute studies in subjects consuming fortified food and supplements. Am. J. Clin. Nutr. 1997, 65, 1790–1795. [Google Scholar] [CrossRef]
- Reynolds, E.H. What is the safe upper intake level of folic acid for the nervous system? Implications for folic acid fortification policies. Eur. J. Clin. Nutr. 2016, 70, 537–540. [Google Scholar] [CrossRef]
- Reeves, P.G. Components of the AIN-93 diets as improvements in the AIN-76A diet. J. Nutr. 1997, 127, 838S–841S. [Google Scholar] [CrossRef]
- Bahous, R.H.; Jadavji, N.M.; Deng, L.; Cosín-Tomás, M.; Lu, J.; Malysheva, O.; Leung, K.-Y.; Ho, M.-K.; Pallàs, M.; Kaliman, P.; et al. High dietary folate in pregnant mice leads to pseudo-MTHFR deficiency and altered methyl metabolism, with embryonic growth delay and short-term memory impairment in offspring. Hum. Mol. Genet. 2017, 26, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Cosín-Tomás, M.; Luan, Y.; Leclerc, D.; Malysheva, O.V.; Lauzon, N.; Bahous, R.H.; Christensen, K.E.; Caudill, M.A.; Rozen, R. Moderate Folic Acid Supplementation in Pregnant Mice Results in Behavioral Alterations in Offspring with Sex-Specific Changes in Methyl Metabolism. Nutrients 2020, 12, 1716. [Google Scholar] [CrossRef]
- Luan, Y.; Leclerc, D.; Cosín-Tomás, M.; Malysheva, O.V.; Wasek, B.; Bottiglieri, T.; Caudill, M.A.; Rozen, R. Moderate Folic Acid Supplementation in Pregnant Mice Results in Altered Methyl Metabolism and in Sex-Specific Placental Transcription Changes. Mol. Nutr. Food Res. 2021, 65, e2100197. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.G.; Daubner, S.C. Modulation of methylenetetrahydrofolate reductase activity by S-adenosylmethionine and by dihydrofolate and its polyglutamate analogues. Adv. Enzym. Regul. 1982, 20, 123–131. [Google Scholar] [CrossRef]
- Whitehead, V.M.; Kamen, B.A.; Beaulieu, D. Levels of Dihydrofolate Reductase in Livers of Birds, Animals, Primates, and Man. Cancer Drug Deliv. 1987, 4, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Cornet, D.; Clement, A.; Clement, P.; Menezo, Y. High doses of folic acid induce a pseudo-methylenetetrahydrofolate syndrome. SAGE Open Med. Case Rep. 2019, 7, 2050313X19850435. [Google Scholar] [CrossRef]
- Ortbauer, M.; Ripper, D.; Fuhrmann, T.; Lassi, M.; Auernigg-Haselmaier, S.; Stiegler, C.; König, J. Folate deficiency and over-supplementation causes impaired folate metabolism: Regulation and adaptation mechanisms in Caenorhabditis elegans. Mol. Nutr. Food Res. 2016, 60, 949–956. [Google Scholar] [CrossRef]
- Koseki, K.; Maekawa, Y.; Bito, T.; Yabuta, Y.; Watanabe, F. High-dose folic acid supplementation results in significant accumulation of unmetabolized homocysteine, leading to severe oxidative stress in Caenorhabditis elegans. Redox Biol. 2020, 37, 101724. [Google Scholar] [CrossRef]
- Ueland, P.M.; Holm, P.I.; Hustad, S. Betaine: A key modulator of one-carbon metabolism and homocysteine status. Clin. Chem. Lab. Med. 2005, 43, 1069–1075. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Jacobs, R.L.; Stead, L.M.; Brosnan, M.E. Methylation demand: A key determinant of homocysteine metabolism. Acta Biochim. Pol. 2004, 51, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Sauer, J.; Mason, J.B.; Choi, S.-W. Too much folate: A risk factor for cancer and cardiovascular disease? Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, R.; Parrott, S.J.; Raj, S.; Cullum-Dugan, D.; Lucus, D. How prevalent is vitamin B12 deficiency among vegetarians? Nutr. Rev. 2013, 71, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, F. Vitamin B12 sources and bioavailability. Exp. Biol. Med. 2007, 232, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Krishnaveni, G.V.; Hill, J.C.; Veena, S.R.; Bhat, D.S.; Wills, A.K.; Karat, C.L.S.; Yajnik, C.S.; Fall, C.H.D. Low plasma vitamin B12 in pregnancy is associated with gestational ‘diabesity’ and later diabetes. Diabetologia 2009, 52, 2350–2358. [Google Scholar] [CrossRef]
- Lai, J.S.; Pang, W.W.; Cai, S.; Lee, Y.S.; Chan, J.K.Y.; Shek, L.P.C.; Yap, F.K.P.; Tan, K.H.; Godfrey, K.M.; van Dam, R.M.; et al. High folate and low vitamin B12 status during pregnancy is associated with gestational diabetes mellitus. Clin. Nutr. 2018, 37, 940–947. [Google Scholar] [CrossRef]
- Radzicka, S.; Ziolkowska, K.; Zaborowski, M.; Brazert, J.; Pietryga, M. Serum homocysteine and vitamin B12 levels in women with gestational diabetes mellitus. Ginekologia Polska 2019, 90, 381–387. [Google Scholar] [CrossRef]
- Paul, L.; Selhub, J. Interaction between excess folate and low vitamin B12 status. Mol. Asp. Med. 2017, 53, 43–47. [Google Scholar] [CrossRef]
- Sukumar, N.; Venkataraman, H.; Wilson, S.; Goljan, I.; Selvamoni, S.; Patel, V.; Saravanan, P. Vitamin B12 Status among Pregnant Women in the UK and Its Association with Obesity and Gestational Diabetes. Nutrients 2016, 8, 768. [Google Scholar] [CrossRef]
- Russell, M.A.; Carpenter, M.W.; Coustan, D.R. Screening and diagnosis of gestational diabetes mellitus. Clin. Obstet. Gynecol. 2007, 50, 949–958. [Google Scholar] [CrossRef]
- Hermann, A.; Sitdikova, G. Homocysteine: Biochemistry, Molecular Biology and Role in Disease. Biomolecules 2021, 11, 737. [Google Scholar] [CrossRef]
- van der Windt, M.; Schoenmakers, S.; van Rijn, B.; Galjaard, S.; Steegers-Theunissen, R.; van Rossem, L. Epidemiology and (Patho)Physiology of Folic Acid Supplement Use in Obese Women before and during Pregnancy. Nutrients 2021, 13, 331. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Deng, H.Y.; Qiao, Z.Y.; Gong, F.X. Homocysteine level and gestational diabetes mellitus: A systematic review and meta-analysis. Gynecol. Endocrinol. 2021, 37, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Tarim, E.; Bagis, T.; Kilicdag, E.; Erkanli, S.; Aslan, E.; Sezgin, N.; Kuscu, E. Elevated plasma homocysteine levels in gestational diabetes mellitus. Acta Obstet. Gynecol. Scand. 2004, 83, 543–547. [Google Scholar] [CrossRef]
- Guven, M.A.; Kilinc, M.; Batukan, C.; Ekerbicer, H.C.; Aksu, T. Elevated second trimester serum homocysteine levels in women with gestational diabetes mellitus. Arch. Gynecol. Obstet. 2006, 274, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Seghieri, G.; Breschi, M.C.; Anichini, R.; De Bellis, A.; Alviggi, L.; Maida, I.; Franconi, F. Serum homocysteine levels are increased in women with gestational diabetes mellitus. Metabolism 2003, 52, 720–723. [Google Scholar] [CrossRef]
- Tarim, E.; Yigit, F.; Kilicdag, E.; Bagis, T.; Demircan, S.; Simsek, E.; Haydardedeoglu, B.; Yanik, F. Early onset of subclinical atherosclerosis in women with gestational diabetes mellitus. Ultrasound Obstet. Gynecol. 2006, 27, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Davari-Tanha, F.; Khan-Mohamadi, F.; Kaveh, M.; Shariat, M. Homocysteine in gestational diabetes and normal pregnancy plus effects of folic acid. Iran. J. Public Health 2008, 37, 118–126. [Google Scholar]
- Atay, A.E.; Simsek, H.; Demir, B.; Sakar, M.N.; Kaya, M.; Pasa, S.; Demir, S.; Sit, D. Noninvasive assessment of subclinical atherosclerosis in normotensive gravidae with gestational diabetes. Herz 2014, 39, 627–632. [Google Scholar] [CrossRef]
- Deng, M.; Zhou, J.; Tang, Z.; Xiang, J.; Yi, J.; Peng, Y.; Di, L.; Zhai, X.; Yang, M.; Du, Y. The correlation between plasma total homocysteine level and gestational diabetes mellitus in a Chinese Han population. Sci. Rep. 2020, 10, 18679. [Google Scholar] [CrossRef]
- Idzior-Waluś, B.; Cyganek, K.; Sztefko, K.; Seghieri, G.; Breschi, M.C.; Waluś-Miarka, M.; Kawalec, E.; Seretny, M.; Sieradzki, J. Total plasma homocysteine correlates in women with gestational diabetes. Arch. Gynecol. Obstet. 2008, 278, 309–313. [Google Scholar] [CrossRef]
- Walker, M.C.; Smith, G.N.; Perkins, S.L.; Keely, E.J.; Garner, P.R. Changes in homocysteine levels during normal pregnancy. Am. J. Obstet. Gynecol. 1999, 180, 660–664. [Google Scholar] [CrossRef]
- López-Quesada, E.; Antònia Vilaseca, M.; Gómez, E.; Lailla, J.M. Are plasma total homocysteine and other amino acids associated with glucose intolerance in uncomplicated pregnancies and preeclampsia? Eur. J. Obstet. Gynecol. Reprod. Biol. 2005, 119, 36–41. [Google Scholar] [CrossRef]
- Metzger, B.E.; Buchanan, T.A.; Coustan, D.R.; de Leiva, A.; Dunger, D.B.; Hadden, D.R.; Hod, M.; Kitzmiller, J.L.; Kjos, S.L.; Oats, J.N.; et al. Summary and Recommendations of the Fifth International Workshop-Conference on Gestational Diabetes Mellitus. Diabetes Care 2007, 30, S251–S260. [Google Scholar] [CrossRef] [PubMed]
- Akturk, M.; Altinova, A.; Mert, I.; Dincel, A.; Sargin, A.; Buyukkagnici, U.; Arslan, M.; Danisman, N. Asymmetric dimethylarginine concentrations are elevated in women with gestational diabetes. Endocrine 2010, 38, 134–141. [Google Scholar] [CrossRef]
- Mascarenhas, M.; Habeebullah, S.; Sridhar, M.G. Revisiting the role of first trimester homocysteine as an index of maternal and fetal outcome. J. Pregnancy 2014, 2014, 123024. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.; Garg, A.; Surana, V.; Aggarwal, S.; Gupta, G.; Singla, S. Vitamin B12 Deficiency is Endemic in Indian Population: A Perspective from North India. Indian J. Endocrinol. Metab. 2019, 23, 211–214. [Google Scholar] [CrossRef]
- Campbell, J.E.; Newgard, C.B. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 2021, 22, 142–158. [Google Scholar] [CrossRef]
- Lain, K.Y.; Catalano, P.M. Metabolic changes in pregnancy. Clin. Obstet. Gynecol. 2007, 50, 938–948. [Google Scholar] [CrossRef]
- Hill, D.J. Placental control of metabolic adaptations in the mother for an optimal pregnancy outcome. What goes wrong in gestational diabetes? Placenta 2018, 69, 162–168. [Google Scholar] [CrossRef]
- Simpson, S.; Smith, L.; Bowe, J. Placental peptides regulating islet adaptation to pregnancy: Clinical potential in gestational diabetes mellitus. Curr. Opin. Pharmacol. 2018, 43, 59–65. [Google Scholar] [CrossRef]
- Patterson, S.; Flatt, P.R.; Brennan, L.; Newsholme, P.; McClenaghan, N.H. Detrimental actions of metabolic syndrome risk factor, homocysteine, on pancreatic β-cell glucose metabolism and insulin secretion. J. Endocrinol. 2006, 189, 301–310. [Google Scholar] [CrossRef]
- Patterson, S.; Scullion, S.M.J.; McCluskey, J.T.; Flatt, P.R.; McClenaghan, N.H. Prolonged exposure to homocysteine results in diminished but reversible pancreatic β-cell responsiveness to insulinotropic agents. Diabetes Metab. Res. Rev. 2007, 23, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Ding, S.; Zhou, L.; Wen, S.; Du, A.; Diao, J. Association between plasma homocysteine levels and pancreatic islet beta-cell function in the patients with type 2 diabetes mellitus: A cross-sectional study from China. Ann. Palliat. Med. 2021, 10, 8169–8179. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Tian, F.-J.; Lin, Y.; Xu, W.-M. Oxidative Stress: Placenta Function and Dysfunction. Am. J. Reprod. Immunol. 2016, 76, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; van Goor, H.; Hillebrands, J.L. Oxidative stress in placental pathology. Placenta 2018, 69, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.N.; Liang, Y.R.; Lin, W.B.; Yao, Z.R.; Chen, D.B.; Chen, P.S.; Ouyang, J. Homocysteine induced oxidative stress in human umbilical vein endothelial cells via regulating methylation of SORBS1. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6948–6958. [Google Scholar] [CrossRef]
- Topal, G.; Brunet, A.; Millanvoye, E.; Boucher, J.L.; Rendu, F.; Devynck, M.A.; David-Dufilho, M. Homocysteine induces oxidative stress by uncoupling of NO synthase activity through reduction of tetrahydrobiopterin. Free. Radic. Biol. Med. 2004, 36, 1532–1541. [Google Scholar] [CrossRef]
- Currò, M.; Gugliandolo, A.; Gangemi, C.; Risitano, R.; Ientile, R.; Caccamo, D. Toxic Effects of Mildly Elevated Homocysteine Concentrations in Neuronal-Like Cells. Neurochem. Res. 2014, 39, 1485–1495. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; De Santo, N.G. Homocysteine and oxidative stress. Amino Acids 2003, 25, 409–417. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8, a006072. [Google Scholar] [CrossRef] [PubMed]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Carletti, J.V.; Correia-Branco, A.; Silva, C.R.; Andrade, N.; Silva, L.O.P.; Martel, F. The effect of oxidative stress induced by tert-butylhydroperoxide under distinct folic acid conditions: An in vitro study using cultured human trophoblast-derived cells. Reprod. Toxicol. 2018, 77, 33–42. [Google Scholar] [CrossRef]
- Jaya, B.; Renuka Devi, M.R.; Karthikeyan, E.; Saikumar, P. A comparative study of oxidative stress among gestational diabetics and normal pregnancy. Natl. J. Physiol. Pharm. Pharmacol. 2019, 9, 86–89. [Google Scholar] [CrossRef]
- Karacay, Ö.; Sepici-Dincel, A.; Karcaaltincaba, D.; Sahin, D.; Yalvaç, S.; Akyol, M.; Kandemir, Ö.; Altan, N. A quantitative evaluation of total antioxidant status and oxidative stress markers in preeclampsia and gestational diabetic patients in 24–36 weeks of gestation. Diabetes Res. Clin. Pract. 2010, 89, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Rueangdetnarong, H.; Sekararithi, R.; Jaiwongkam, T.; Kumfu, S.; Chattipakorn, N.; Tongsong, T.; Jatavan, P. Comparisons of the oxidative stress biomarkers levels in gestational diabetes mellitus (GDM) and non-GDM among Thai population: Cohort study. Endocr. Connect. 2018, 7, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yang, H.; Geng, Q.; Ma, Q.; Long, Y.; Zhou, C.; Chen, M. Association of oxidative stress biomarkers with gestational diabetes mellitus in pregnant women: A case-control study. PLoS ONE 2015, 10, e0126490. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, T.; Hu, S.; Sun, L. Association of serum lipid peroxidation and glutathione peroxidase 4 levels with clinical outcomes and metabolic abnormalities among patients with gestational diabetes mellitus: A case-control study in the Chinese population. Front. Biosci. 2022, 27, 68. [Google Scholar] [CrossRef]
- Lewandowski, K.C.; Stojanovic, N.; Press, M.; Tuck, S.; Lewiński, A.; Karbownik-Lewińska, M. Raised concentrations of lipid peroxidation products (LPO) in pregnant women with impaired glucose tolerance. Ann. Agric. Environ. Med. 2014, 21, 429–434. [Google Scholar] [CrossRef]
- Di Simone, N.; Maggiano, N.; Caliandro, D.; Riccardi, P.; Evangelista, A.; Carducci, B.; Caruso, A. Homocysteine Induces Trophoblast Cell Death with Apoptotic Features1. Biol. Reprod. 2003, 69, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Di Simone, N.; Riccardi, P.; Maggiano, N.; Piacentani, A.; D’Asta, M.; Capelli, A.; Caruso, A. Effect of folic acid on homocysteine-induced trophoblast apoptosis. Mol. Hum. Reprod. 2004, 10, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Kamudhamas, A.; Pang, L.; Smith, S.D.; Sadovsky, Y.; Nelson, D.M. Homocysteine thiolactone induces apoptosis in cultured human trophoblasts: A mechanism for homocysteine-mediated placental dysfunction? Am. J. Obstet. Gynecol. 2004, 191, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Sgarbosa, F.; Barbisan, L.F.; Brasil, M.A.; Costa, E.; Calderon, I.M.; Gonçalves, C.R.; Bevilacqua, E.; Rudge, M.V. Changes in apoptosis and Bcl-2 expression in human hyperglycemic, term placental trophoblast. Diabetes Res. Clin. Pract. 2006, 73, 143–149. [Google Scholar] [CrossRef]
- Araújo, J.R.; Correia-Branco, A.; Ramalho, C.; Keating, E.; Martel, F. Gestational diabetes mellitus decreases placental uptake of long-chain polyunsaturated fatty acids: Involvement of long-chain acyl-CoA synthetase. J. Nutr. Biochem. 2013, 24, 1741–1750. [Google Scholar] [CrossRef]
- Belkacemi, L.; Kjos, S.; Nelson, D.M.; Desai, M.; Ross, M.G. Reduced apoptosis in term placentas from gestational diabetic pregnancies. J. Dev. Orig. Health Dis. 2013, 4, 256–265. [Google Scholar] [CrossRef]
- Nwabuobi, C.; Arlier, S.; Schatz, F.; Guzeloglu-Kayisli, O.; Lockwood, C.J.; Kayisli, U.A. hCG: Biological Functions and Clinical Applications. Int J. Mol. Sci 2017, 18, 2037. [Google Scholar] [CrossRef]
- Donovan, B.M.; Nidey, N.L.; Jasper, E.A.; Robinson, J.G.; Bao, W.; Saftlas, A.F.; Ryckman, K.K. First trimester prenatal screening biomarkers and gestational diabetes mellitus: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0201319. [Google Scholar] [CrossRef]
- Ahmed, T.; Fellus, I.; Gaudet, J.; MacFarlane, A.J.; Fontaine-Bisson, B.; Bainbridge, S.A. Effect of folic acid on human trophoblast health and function in vitro. Placenta 2016, 37, 7–15. [Google Scholar] [CrossRef]
- Rush, E.C.; Katre, P.; Yajnik, C.S. Vitamin B12: One carbon metabolism, fetal growth and programming for chronic disease. Eur. J. Clin. Nutr. 2014, 68, 2–7. [Google Scholar] [CrossRef]
- Sobczyńska-Malefora, A.; Harrington, D.J. Laboratory assessment of folate (vitamin B9) status. J. Clin. Pathol. 2018, 71, 949. [Google Scholar] [CrossRef] [PubMed]
- McMullin, M.F.; Young, P.B.; Bailie, K.E.M.; Savage, G.A.; Lappin, T.R.J.; White, R. Homocysteine and methylmalonic acid as indicators of folate and vitamin B12 deficiency in pregnancy. Clin. Lab. Haematol. 2001, 23, 161–165. [Google Scholar] [CrossRef]
- Shah, T.; Joshi, K.; Mishra, S.; Otiv, S.; Kumbar, V. Molecular and cellular effects of vitamin B12 forms on human trophoblast cells in presence of excessive folate. Biomed. Pharmacother. 2016, 84, 526–534. [Google Scholar] [CrossRef]
- Selhub, J.; Morris, M.S.; Jacques, P.F. In vitamin B12 deficiency, higher serum folate is associated with increased total homocysteine and methylmalonic acid concentrations. Proc. Natl. Acad. Sci. USA 2007, 104, 19995–20000. [Google Scholar] [CrossRef]
- Wiedeman, A.M.; Barr, S.I.; Green, T.J.; Xu, Z.; Innis, S.M.; Kitts, D.D. Dietary Choline Intake: Current State of Knowledge across the Life Cycle. Nutrients 2018, 10, 1513. [Google Scholar] [CrossRef]
- Friso, S.; Udali, S.; De Santis, D.; Choi, S.-W. One-carbon metabolism and epigenetics. Mol. Asp. Med. 2017, 54, 28–36. [Google Scholar] [CrossRef]
- Ganz, A.B.; Shields, K.; Fomin, V.G.; Lopez, Y.S.; Mohan, S.; Lovesky, J.; Chuang, J.C.; Ganti, A.; Carrier, B.; Yan, J.; et al. Genetic impairments in folate enzymes increase dependence on dietary choline for phosphatidylcholine production at the expense of betaine synthesis. FASEB J. 2016, 30, 3321–3333. [Google Scholar] [CrossRef]
- Huo, X.; Li, J.; Cao, Y.-F.; Li, S.-N.; Shao, P.; Leng, J.; Li, W.; Liu, J.; Yang, K.; Ma, R.C.W.; et al. Trimethylamine N-Oxide Metabolites in Early Pregnancy and Risk of Gestational Diabetes: A Nested Case-Control Study. J. Clin. Endocrinol. Metab. 2019, 104, 5529–5539. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Du, Y.; Li, X.; Yang, J.; Zhang, X.; Wei, Y.; Zhao, Y. Maternal Plasma Betaine in Middle Pregnancy Was Associated with Decreased Risk of GDM in Twin Pregnancy: A Cohort Study. Diabetes Metab. Syndr. Obes. 2021, 14, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Barzilay, E.; Moon, A.; Plumptre, L.; Masih, S.P.; Sohn, K.-J.; Visentin, C.E.; Ly, A.; Malysheva, O.; Croxford, R.; Caudill, M.A.; et al. Fetal one-carbon nutrient concentrations may be affected by gestational diabetes. Nutr. Res. 2018, 55, 57–64. [Google Scholar] [CrossRef]
- King, J.H.; Kwan, S.T.C.; Yan, J.; Jiang, X.; Fomin, V.G.; Levine, S.P.; Wei, E.; Roberson, M.S.; Caudill, M.A. Maternal Choline Supplementation Modulates Placental Markers of Inflammation, Angiogenesis, and Apoptosis in a Mouse Model of Placental Insufficiency. Nutrients 2019, 11, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvi, F.; Gadda, G. Human choline dehydrogenase: Medical promises and biochemical challenges. Arch. Biochem. Biophys. 2013, 537, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Sauceda, C.; Webster, N.J.G. Mitochondrial Dysfunction in Obesity and Reproduction. Endocrinology. 2021, 162, bqaa158. [Google Scholar] [CrossRef] [PubMed]
- Abbade, J.; Klemetti, M.M.; Farrell, A.; Ermini, L.; Gillmore, T.; Sallais, J.; Tagliaferro, A.; Post, M.; Caniggia, I. Increased placental mitochondrial fusion in gestational diabetes mellitus: An adaptive mechanism to optimize feto-placental metabolic homeostasis? BMJ DRC. 2020, 8, e000923. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Authors | Study Country | Pregnancy Status (n) | Weeks’ Gestation | Measure | Results |
|---|---|---|---|---|---|
| Zhu et al., 2016 [7] | China | Non-GDM (1689) vs. GDM (249) | <12 | FA supplementation | aOR: 2.25 95% CI: 1.35–3.76 |
| Cheng et al., 2019 [9] | China | Non-GDM (853) vs. GDM (97) | ≥3 months pre-conception | FA supplementation | aRR: 1.72 95% CI: 1.17–2.53, p < 0.01 |
| Huang et al., 2019 [10] | China | Non-GDM (293) vs. GDM (33) | 16–18 | FA supplementation | aOR: 3.45 95% CI: 1.01–11.8, p < 0.05 |
| Chen et al., 2021 [8] | China | Non-GDM (878) vs. GDM (180) | 9–13 | FA supplementation | aOR: 1.73 95% CI: 1.19–2.53, p = 0.004 |
| RBC Folate | aOR: 1.58 95% CI: 1.03–2.41, p = 0.033 | ||||
| Xie et al., 2019 [11] | China | Uncomplicated (1890) vs. GDM (392) | 19–24 | RBC folate | RR per 1-SD increase: 1.16 95% CI 1.03–1.30, p = 0.012 |
| Liu et al., 2020 [12] | China | Non-GDM (299) vs. GDM (67 | <12 | RBC Folate | aOR: 2.473 95% CI: 1.013–6.037, p = 0.047 |
| Li et al., 2019 [14] | China | Uncomplicated (316) vs. GDM (90) | 24–28 | Serum folate | OR: 1.98 95% CI: 1.00–3.90, p = 0.049 |
| Saravanan et al., 2021 [15] | UK | Uncomplicated (3702) vs. GDM (526) | 12.5 ± 1.4 | Serum folate | aRR: 1.11 95% CI: 1.036–1.182, p = 0.002 |
| Jankovic-Karasoulos et al., 2021 [16] | Australia and New Zealand | Uncomplicated (111) vs. GDM (33) | 15 ± 1 | Serum folate | mean ± SD (nmol/L): 31.9 ± 11.2 vs. 37.6 ± 8.0, p = 0.007) aOR: 1.22 (0.93–1.59), p = 0.149 |
| Authors | Study Country | Pregnancy Status (n) | Weeks’ Gestation | Measure | Results |
|---|---|---|---|---|---|
| Sukumar et al., 2016 [115] | UK | B12-deficient < 150 pmol/L (90) vs. B12-replete > 150 pmol/L (254) | 26.9 ± 5.3 | WHO 1999 GDM criteria | OR: 2.59 95% CI 1.35–4.98, p = 0.004. aOR: 2.05, 95% CI: 1.03–4.10, p = 0.04 |
| Saravan et al., 2021 [15] | UK | B12-deficient < 220 pmol/L (1790) vs. B12-replete > 220 pmol/L (2530) | 12.5 ± 1.4 | IADPSG-GDM | aRR: 1.383, 95% CI 1.157–1.652, p = 0.0004 |
| Uncomplicated (3687) vs. GDM (633) | 12.5 ± 1.4 | Serum B12 | aRR: 0.856, 95% CI: 0.786–0.933, p = 0.0004 | ||
| B12 tertile 1 + folate tertile 3 | aRR: 1.742 95% CI: 1.226–2.437, p = 0.003 | ||||
| Li et al., 2019 [14] | China | Uncomplicated (110) vs. GDM (27) | 24–28 | Serum folate:B12 ratio 26.67–41.03 | aOR: 1.53 95% CI: 0.79–2.97, p = 0.211 |
| Uncomplicated (93) vs. GDM (43) | 24–28 | Serum folate:B12 ratio ≥ 41.03 | aOR: 3.08 95% CI: 1.63–5.83, p = 0.001 | ||
| Lai et al., 2018 [112] | Singapore | Folate Tertile 1 (Ref) (193) vs. Folate Tertile 2 (164) | 26–28 | WHO 1999 GDM criteria | aOR: 1.94 95% CI: 1.04–3.62, p = 0.036 |
| Folate Tertile 1 (Ref) (193) vs. Folate Tertile 3 (156) | 26–28 | WHO 1999 GDM criteria | aOR: 1.97 95% CI: 1.05–3.68, p = 0.034 | ||
| Krishnaveni et al., 2009 [111] | India | Folate ≤ 21.3 nmol/L (129) vs. Folate > 21.3–45.4 nmol/L (114) and Folate > 45.4 nmol/L (91) | 30 | GDM Carpenter–Coustan criteria [116] | 5.4%, 10.5%, 10.9% (Tertile 1, 2, and 3, respectively), p = 0.04 |
| Authors | Study Country | Pregnancy Status (n) | Weeks’ Gestation | Hcy Measure | Results |
|---|---|---|---|---|---|
| Tarim et al., 2004 [120] | Turkey | Normoglycemic ≤ 7.5 nmol/L, 1 h-50 g glucose (210) vs. glucose intolerant, >7.5 nmol/L glucose challenge, normal oGTT (66) vs. GDM (28) | 24–28 | Plasma 8 h fasting | Mean ± SD (μmol/L) Group 1: 4.80 ± 0.98 Group 2: 5.51 ± 1.08 Group 3: 5.70 ± 0.90 (p < 0.001) |
| Guven et al., 2006 [121] | Turkey | Normoglycemic ≤ 7.8 nmol/L, 1 h-50 g glucose (147) vs. glucose intolerant > 7.8 nmol/L glucose challenge, normal oGTT (46) vs. GDM (30) | 24–28 | Serum | Mean ± SD (μmol/L) Group 1: 7.4 ± 1.6 Group 2: 8.1 ± 2.5 Group 3: 9.0 ± 3.1, p < 0.01 |
| Seghieri et al., 2003 [122] | Italy | Non-GDM (78) vs. GDM (15) | 24–28 | Serum | Mean ± SD (μmol/L) Control: 4.45 ± 1.52 GDM: 5.88 ± 2.26, p = 0.003 |
| Tarim et al., 2006 [123] | Turkey | Non-GDM (40) vs. GDM (30) | 24–28 | Plasma | Mean ± SD (μmol/L) Control: 5.03 ± 0.91 GDM: 5.96 ± 1.70 p = 0.027 |
| Davari-Tanha et al., 2008 [124] | Iran | Non-GDM (40) vs. GDM (40) | 24–28 | Plasma 8 h fasting | Mean ± SD (μmol/L) Control: 5.05 ± 1.1 GDM: 7.8 ± 1.6 p < 0.0001 |
| Atay et al., 2014 [125] | NS | Uncomplicated (38) vs. GDM (37) | 24–28 | Serum 12 h fasting | Mean ± SD mmol/l) Control: 5.91 ± 3.87 GDM: 9.57 ± 4.46 p < 0.001 |
| Deng et al., 2020 [126] | China | Non-GDM (350) vs. GDM (346) | 24–28 | Plasma | Mean ± SD (μmol/L) Control: 6.17 ± 1.29 GDM: 6.61 ± 1.32 p = 0.001 |
| Idzior-Waluś et al., 2008 [127] | Poland | Non-GDM (17) vs. GDM (44) | 26–32 | Serum | Mean ± SD (μmol/L) Control: 7.4 ± 1.1 GDM 8 ± 2.0 NS |
| Radzicka et al., 2019 [113] | Poland | Uncomplicated (19) vs. GDM (60) | 24–28 | Serum | Mean ± IQR (μmol/L) Control: 8.02 ± 2.27 GDM: 7.41 ± 2.61 (NS) |
| López-Quesada et al., 2005 [129] | Spain | Normoglycemic ≤ 7.8 nmol/L, 1 h-50 g glucose (190) vs. Glucose intolerant (18) > 7.8 nmol/L glucose challenge, normal oGTT vs. GDM (17) | 34 | Plasma fasting | Median ± SD (μmol/L) Group 1: 6.6 ± 2.0 Group 2: 5.0 ± 1.7 Group 3 6.8 ± 2.7 |
| Akturk et al., 2010 [131] | Turkey | Normoglycemic (69) vs. GDM (54) | 32–39 | Plasma | Mean ± SEM (μmol/L) Control: 5.62 ± 0.34 GDM: 5.20 ± 0.30 |
| Mascarenhas et al. 2014 [132] | India | Normoglycemic (83) vs. GDM (7) | 8–12 | Serum overnight fasting | Mean (μmol/L) Control: 14.41 ± 7.98 GDM: 15.66 ± 7.61 p = 0.6312 |
| Authors | Study Country | Pregnancy Status (n) | Weeks’ Gestation | Measure | Results |
|---|---|---|---|---|---|
| Huo et al., 2019 [175] | China | Uncomplicated (243) vs. GDM (243) | Median: 10 (IQR: 9–11) | Serum betaine | Mean (IQR) (nmol/mL) Control: 290.4 (244.2–378.8) GDM: 229.7 (195.6–279.9), p < 0.0001 |
| Betaine ≤ 200 nmol/mL (90) vs. Betaine > 200 nmol/mL (396) | Median: 10 (IQR: 9–11) | GDM (WHO 2013 criteria) | OR: 5.00 95% CI: 2.76–9.07, p < 0.0001 aOR: 4.88 95% CI 2.51–9.50, p < 0.0001 | ||
| Gong et al., 2021 [176] | China | Betaine Tertile 1 (62) vs. Betaine Tertile 2 (63) vs. Betaine Tertile 3 (62) | 5.4–11.4 | IADPSG-GDM | aRR: 0.41 (95% CI: 0.19– 0.86, p-trend = 0.015 |
| Barzilay et al., 2018 [177] | Canada | Uncomplicated (296) vs. GDM (18) | 12–16 | Plasma betaine | Mean ± SD (μmol/L): 13.4 ± 4.1 vs. 12.1 ± 2.4, p = 0.15 |
| Uncomplicated (278) vs. GDM (16) | 37–42 | Plasma betaine | Mean ± SD (μmol/L): 10.4 ± 2.8 vs. 10.3 ± 2.2, p = 0.92 | ||
| Uncomplicated (252) vs. GDM (14) | 28–42 | Cord blood plasma betaine | Mean ± SD (μmol/L): 21.2 ± 4.7 vs. 18.5 ± 3.9, p = 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williamson, J.M.; Arthurs, A.L.; Smith, M.D.; Roberts, C.T.; Jankovic-Karasoulos, T. High Folate, Perturbed One-Carbon Metabolism and Gestational Diabetes Mellitus. Nutrients 2022, 14, 3930. https://doi.org/10.3390/nu14193930
Williamson JM, Arthurs AL, Smith MD, Roberts CT, Jankovic-Karasoulos T. High Folate, Perturbed One-Carbon Metabolism and Gestational Diabetes Mellitus. Nutrients. 2022; 14(19):3930. https://doi.org/10.3390/nu14193930
Chicago/Turabian StyleWilliamson, Jessica M., Anya L. Arthurs, Melanie D. Smith, Claire T. Roberts, and Tanja Jankovic-Karasoulos. 2022. "High Folate, Perturbed One-Carbon Metabolism and Gestational Diabetes Mellitus" Nutrients 14, no. 19: 3930. https://doi.org/10.3390/nu14193930